Article Text

Abstract
Purpose Inherited peripheral neuropathies (IPN) represent a large heterogenous group of hereditary diseases with more than 100 causative genes reported to date. In this context, targeted next-generation sequencing (NGS) offers the opportunity to screen all these genes with high efficiency in order to unravel the genetic basis of the disease. Here, we compare the diagnostic yield of targeted NGS with our previous gene by gene Sanger sequencing strategy. We also describe several novel likely pathogenic variants.
Design and participants We have completed the targeted NGS of 81 IPN genes in a cohort of 123 unrelated patients affected with diverse forms of IPNs, mostly Charcot-Marie-Tooth disease (CMT): 23% CMT1, 52% CMT2, 9% distal hereditary motor neuropathy, 7% hereditary sensory and autonomic neuropathy and 6.5% intermediate CMT.
Results We have solved the molecular diagnosis in 49 of 123 patients (~40%). Among the identified variants, 26 variants were already reported in the literature. In our cohort, the most frequently mutated genes are respectively: MFN2, SH3TC2, GDAP1, NEFL, GAN, KIF5A and AARS. Panel-based NGS was more efficient in familial cases than in sporadic cases (diagnostic yield 49%vs19%, respectively). NGS-based search for copy number variations, allowed the identification of three duplications in three patients and raised the diagnostic yield to 41%. This yield is two times higher than the one obtained previously by gene Sanger sequencing screening. The impact of panel-based NGS screening is particularly important for demyelinating CMT (CMT1) subtypes, for which the success rate reached 87% (36% only for axonal CMT2).
Conclusion NGS allowed to identify causal mutations in a shorter and cost-effective time. Actually, targeted NGS is a well-suited strategy for efficient molecular diagnosis of IPNs. However, NGS leads to the identification of numerous variants of unknown significance, which interpretation requires interdisciplinary collaborations between molecular geneticists, clinicians and (neuro)pathologists.
- next generation sequencing
- copy number variation
- inherited peripheral neuropathies
- mutation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
First study concerning patients with inherited peripheral neuropathies in South of France.
New single-nucleotide variation mutations important for the scientific and medical community.
New copy number variations detected by the analysis of next-generation sequencing data.
Lack of power (123 patients).
Lack of functional validation of variants of unknown significance.
Introduction
We defined three main categories of inherited peripheral neuropathy (IPN): hereditary motor and sensory neuropathy, more commonly called Charcot-Marie-Tooth disease (CMT), hereditary sensory and autonomic neuropathy (HSAN) and distal hereditary motor neuropathy (dHMN).
With a prevalence of 1/2500,1 CMT is one of the most frequent cause of neurological disability, characterised by extensive phenotypic and genetic heterogeneity, with all modes of inheritance described. Based on histopathological and electrophysiological criteria, CMT are further categorised into CMT type 1, or demyelinating type (CMT1/HMSN1), and CMT type 2, or axonal type (CMT2/HMSN2). While CMT type 1 is associated with reduced nerve conduction velocities, correlated to decreased myelination of the peripheral nerves, CMT type 2 is characterised by decreased amplitudes of motor and sensory nerve action potentials, related to primarily axonal loss in the peripheral nerve fibres. Patients with both signs of demyelination and axonal degeneration are diagnosed with intermediate CMT, although there is much controversy about the exact definition of intermediate CMT.2
The disease usually starts in childhood or the teenage years and it generally aggravates slowly and progressively. Clinically, most patients present distal motor and sensory weakness associated with feet deformations (most frequently pes cavus) and sometimes other skeletal deformations such as scoliosis. Additional manifestations may exist such as deafness, optic atrophy or pyramidal signs. The disease is characterised by high clinical heterogeneity, both intrafamilial and interfamilial. Concerning the latter, the severity of the disease is highly variable, ranging from almost asymptomatic adult patients to severely disabled children.
Genetically, the disease is also highly heterogeneous, with more than 100 defective genes reported today.3 4 In our laboratory, before the advent of next-generation sequencing (NGS), the screening strategy was based on Sanger sequencing of candidate genes, except for the PMP22 duplication, following the recommendations of the French Network of Molecular Diagnosis Laboratories for NeuroMuscular Diseases (http://www.anpgm.fr/), who had set up decision trees based on data from the literature. The screening of one specific gene was determined by clinical, genealogical and electrophysiological criteria. For all patients, the 1.5 Mb duplication at chromosome 17p11.2 encompassing the PMP22 gene, the most frequent cause of CMT, was first achieved by multiplex ligation-dependent probe amplification (MLPA). In negative cases, a subsequent gene by gene sequential screening was carried out, and the screened genes were different whether the patient presented demyelinating or axonal CMT. For patients with autosomal-dominant demyelinating CMT (CMT1), the screening of GJB1, the gene encoding connexin 32, was then carried out through Sanger sequencing, followed by the sequential screening of: MPZ, LITAF/SIMPLE, NEFL, GDAP1 and EGR2. In autosomal-dominant axonal CMT (CMT2), the patients were subjected to Sanger sequencing screening of the following genes: GJB1, MFN2 (especially if the patient show pyramidal signs or optic atrophy), then MPZ and NEFL.
In the last few years, the molecular diagnosis improved with the advent of NGS, which is now the strategy used in routine in our laboratory, in patients negative for the PMP22 duplication. The list of genes involved in IPNs is constantly rising, and is now above 100.
Our objective is to compare the diagnostic yield of a targeted NGS strategy (panel of 81 IPN/CMT genes) with the previous gene by gene Sanger sequencing strategy, by comparing the results of NGS in a cohort of 123 patients and the results of Sanger sequencing in a group of 56 patients. We compare the molecular diagnostic resolution rates between demyelinating and axonal CMT forms. We also report on new likely pathogenic variants, not yet described in the literature, and we present the most frequently mutated genes in our group. Finally, we describe two candidate copy number variations (CNVs), identified from the NGS data.
Materials and methods
Clinical data
For NGS screening, we have prospectively included 123 index cases affected with hereditary motor and sensory neuropathy, dHMN and HSAN, seen in consultation in the Neuromuscular Disease Reference Center, since 2015.
For Sanger sequencing, we have studied a retrospective cohort of 56 patients seen in consultation between 2012 and 2014.
Patient and public involvement
Patients and/or public were involved neither in the design of this study protocol nor in the development of the research question. Patients and/or public will not be involved in the recruitment process. The results of this study will be presented at the next FILNEMUS conference.
Statistical analysis
We have compared the targeted NGS strategy to the previous gene analysis by Sanger sequencing on a retrospective cohort of 56 patients seen between 2012 and 2014 using a two-failed Fisher’s exact test.
Genetic studies
Samples
A written and signed approval has been collected from the patients in accordance with French recommendations as well as in agreement with the local ethics committee rules.
DNA was extracted from peripheral blood using standard procedures. DNAs were prepared and stored at the accredited Biological Resource Centre (CRB TAC component (NF S96-900 and ISO 9001 v2015 Certification) Department of Medical Genetics, Timone Hospital of Marseille (APHM). All DNAs belong to a biological sample collection declared to the French Ministry of Health (declaration number DC-2008–429) whose use for research purposes was authorised by the French ministry of Health (authorisation number AC-2011–1312 and AC-2017–2986).
All patients were searched for the PMP22 duplication before including them in the NGS analysis protocol. To test for the PMP22 duplication, we used an MLPA protocol (MRC-Holland) following the manufacturer’s recommendations.
Next-generation sequencing
Library preparation and sequencing
Libraries were prepared using the ClearSeq Inherited Disease Panel from Agilent (Santa Clara, California, USA), which enables the capturing of 2742 genes known to cause inherited disorders. The coding regions and flanking intronic regions of the 2742 genes were enriched, in solution, using the SureSelect Target Enrichment System from Agilent (Santa Clara, California, USA), following the manufacturer recommendations.
For sequencing, we used the Ion Proton platform (Thermo Fisher Scientific, USA).
Computational analysis
After capturing enrichment and sequencing, raw data were converted to Fastq files, aligned to the reference sequence of the human genome (University of California Santa Cruz (UCSC) hg19/GRCh37), and annotated, using the Ion Proton platform integrated workflow (Thermo Fisher Scientific, USA). The obtained variant call format and binary alignment map (BAM) files were used for variant search as described below.
In a diagnosis settings, we realised a targeted analysis of the NGS data, focusing on a list of 81 IPN genes (see online supplementary table 1). The data were filtered, using an ‘in-house’ tool for variant annotation and Filtering Variant Annotation and Filter Tool (https://varaft.eu/download.php)5: (1) variants with allele frequencies <1% in the Exome Aggregation Consortium (ExAC) data set (http://exac.broadinstitute.org/) were removed, (2) the remaining variants were filtered based on their type and genomic localisation, thus, synonymous, intronic, variants in intergenic, 3’ and 5’ Untranslated Region (UTR) regions were discarded. In order to predict the deleterious effect of the identified sequence variations, different bioinformatics tools were applied, such as MutationTaster (http://www.mutationtaster.org/),6 Sorting Intolerant From Tolerant (SIFT) (http://sift.bii.a-star.edu.sg/),7 PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)8 and Universal Mutation Database (UMD) predictor (http://umd-predictor.eu/).9
Supplementary file 1
In order to check whether variants had already been inventoried and classified, we looked up in Online Mendelian Inheritance in Man (OMIM) (https://www.ncbi.nlm.nih.gov/omim), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD (http://www.hgmd.cf.ac.uk). Finally, thanks to the American College of Medical Genetics and Genomics (ACMG) recommendations (Richards classification, 2015)10 and to in silico analyses, we classified these variants into five categories: pathogenic, probably pathogenic, variant of unknown significance (VUS), probably benign and benign.
Copy number variations
In order to identify CNV in our cohort, we used ExomeDepth.11 This tool uses the BAM files from the NGS sequencing run as well as the bed file containing the target regions to be studied (81 genes, see online supplementary table 1). This tool allows to compare the reading depth of our patients’ BAM files, to a set of reference samples, in order to eliminate the capturing and sequencing mistakes. Variations in the depth of sequencing are linearly correlated to the variation of the copy number. Deletions and duplications found by ExomeDepth were confirmed by a quantitative (Q)-PCR (Applied Biosystems ViiA 7 Real-Time PCR System).12 The CNV analysis of TFG and FGD4 genes was performed using a commercial kit TaqMan Copy Number Assays (HS00918833-CN) by real-time PCR multiplex. Each analysis was done in triplicate.
Sanger sequencing
Variants found by NGS were confirmed and segregated by Sanger sequencing. In our previous sequential gene by gene analysis, the complete coding sequence of genes were PCR amplified and sequenced by fluorescent Sanger sequencing.
In both situations, genomic and cDNA sequences of the genes were obtained from the UCSC Genomic Browser, February 2009, human reference sequence (GRCh37). Primers used for PCR amplification were designed using Primer3 software (http://frodo.wi.mit.edu) to amplify the region surrounding the candidate DNA variations.
PCR products were purified by mixing with a volume ratio (1/8) (36 uL) of AMPure beads (Beckman Coulter, USA) according to the manufacturer’s instructions and both strands were sequenced using the Big Dye Terminator V.1.1 Cycle Sequencing Kit (Applied Biosystems). Sequence reactions were purified on Sephadex G50 (Amersham Pharmacia Biotech, Foster City, California, USA) and capillary electrophoresis was performed on Genetic Analyser ABI3500XL (Life Technologies, USA). Electrophoregrams were analysed on the Sequence Analysis Software V.5.2 (Applied Biosystems) and aligned with the reference sequence using Sequencher V.5.4.6.
Results
Among the 123 patients, 28 (23%) had CMT type 1, 64 (52%) had CMT type 2, 11 (9%) had dHMN, 9 (7%) had HSAN, 8 (6,5%) had intermediary CMT and 3 (2,5%) had CMT without clinical information. Table 1 sums up the phenotypic features of the patients for whom we identified a mutation or a candidate variant (table 1A for patients with CMT1, table 1B for patients with CMT2 and table 1C for patients with others forms of IPN).
Clinical and electrophysiological features from 19 IPN index patients with CMT1, for whom a candidate variant was identified
Clinical and electrophysiological features from 27 IPN index patients with CMT2, for whom a candidate variant was identified
Clinical and electrophysiological features from 14 IPN index patients with hereditary sensory and autonomic neuropathy (HSAN), distal hereditary motor neuropathy (dHMN) and intermediate CMT, for whom a candidate variant was identified
The average depth obtained in our panel of 81 genes was 196X and the average coverage at 20X was 98%, with a weaker coverage at 20X for SOX10, INF2 and CTDP1 genes (81%, 86% and 86% respectively).
Through targeted NGS of 81 IPN genes (see online supplementary table 1), we found one or several potentially pathogenic variants in 60 patients from our cohort of 123 index cases, thereby defining a success rate of 49%. The average age was 20 for positive cases, while it was significantly higher (45 years old) for negative cases.
More precisely, we found a pathogenic variant for 49 patients (40%) and a potentially pathogenic variant for 11 patients (9%). Thirty-seven per cent of cases showed a recessive transmission and 63% a dominant transmission. Twenty-six variants were reported in the literature, whereas 52 variants were never reported. Among these 52 new variants, we were able to confirm the pathogenicity of 25, based on: (1) their low frequency in the ExAC database, (2) compatible phenotype, (3) segregation in the family study and (4) in silico pathogeneicity prediction. However, we were unable to establish the pathogenicity in 11 patients requiring further explorations at either clinical or genetic levels (see table 2A,B).
Known mutations identified in 81 IPN genes by next-generation sequencing
Novel pathogenic sequence variations identified in IPN genes by next-generation sequencing
Among the 28 patients affected with demyelinating forms, 16 (57%) had a pathogenic variant, 3 (11%) had a potentially pathogenic variant and 9 (32%) remained negative. The most frequently mutated gene was SH3TC2 responsible for CMT4C with five variants found, of which two were not reported in the literature. The other mutations were found in the following genes: NEFL (2), FIG4 (2), KIF1B (2), HK1 (2), INF2 (2), LITAF/SIMPLE (2), DNM2 (1), LMNA (1), DCTN1 (1), GJB1 (2), GDAP1 (1), TRPV4 (1), AARS (1) and LRSAM1 (1).
Among the 64 patients with axonal forms, 23 (36%) had a pathogenic variant, 4 (6%) had a potentially pathogenic variant and 37 (58%) remained negative. The most frequently mutated gene was MFN2 with six variants found, of which two were not reported in the literature, then GAN with four variants found, of which two were not reported, and then the following genes: NEFL (3), GDAP1 (3), AARS (2), IGHMBP2 (2), DCTN1 (1), DARS2 (2), KIF5A (2), INF2 (2), LRSAM1 (1), KIF1B (1), SETP9 (1), ARHGEF10 (1), HSPB1 (1), SPTLC1 (1), BAG3 (1) and BICD2 (1).
Among the 11 patients who had dHMN, 5 (45%) had a pathogenic variant and 6 (55%) remained negative. The most frequently mutated gene was SPG11 with two variants found not described in the literature, and then DYNC1H1 (1), TRPV4 (1), REEP1 (1) and MYH14 (1).
Among the nine patients who had HSAN, three (33%) had a pathogenic variant, two (22%) had a potentially pathogenic variant and four (45%) remained negative. SPTLC2 was the most frequently mutated gene with two variants found, of which one was not reported. Only one variant was reported in the following genes: FAM134B, HSPB1 and NEFL.
Among the eight patients who had intermediate CMT, two (25%) had a pathogenic variant, two (25%) had a potentially pathogenic variant and four (50%) remained negative. A potentially pathogenic variant was found in genes: DNM2, KIF5A, YARS and INF2.
Overall, the most frequently mutated genes were (by decreasing order) (see figure 1): MFN2 (7.7%), SH3TC2 (6.4%), NEFL (5.1%), GDAP1 (5.1%) and GAN (5.1%). Then come the following genes : AARS (3.8%), KIF5A (3.8%), KIF1B (2.6%), INF2 (2.6%), DARS2 (2.6%), DCTN1 (2.6%), DNM2 (2.6%), FIG4 (2.6%), HK1 (2.6%), IGHMBP2 (2.6%), LITAF (2.6%), SPG11 (2.6%), SPTLC2 (2.6%), MYH14 (2.6%), GJB1 (1.3%), HSPB1 (1.3%), TRPV4 (1.3%), LRSAM1 (1.3%), BICD2 (1.3%), SPTLC1 (1.3%), REEP1 (1.3%), HSPB3 (1.3%), FAM134B (1.3%), DYNC1H1 (1.3%) and BAG3 (1.3%).

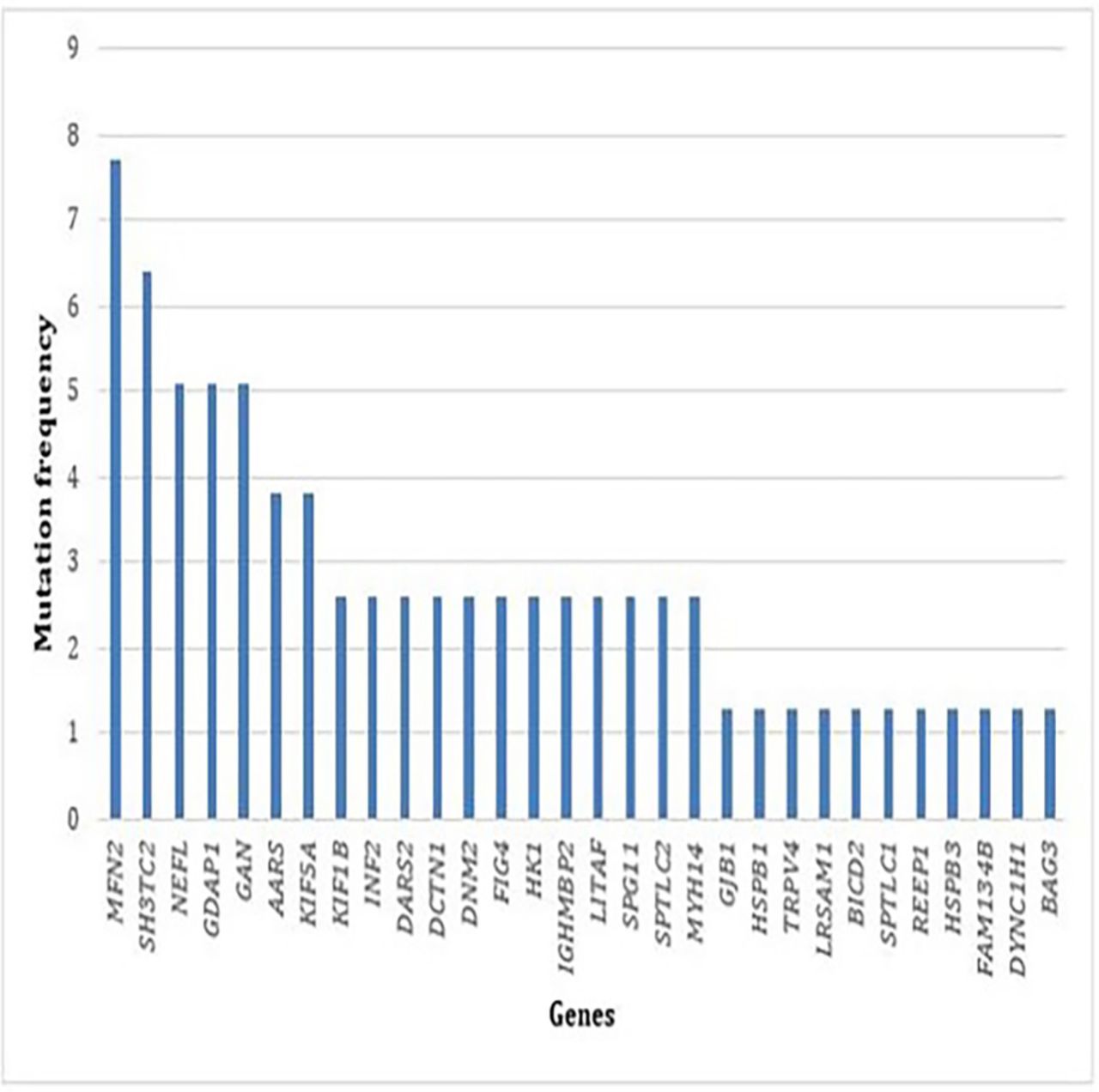{kind=link}
Frequency of mutations in each gene identified in our cohort of 123 patients.
In our laboratory, before the advent of targeted NGS, 20 genes responsible for IPN were explored by Sanger sequencing : MPZ, PMP22, PRX, EGR2, MTMR2, NDRG1, HSPB1, HSPB8, BSCL2, FGD4, LMNA, LITAF, TRPV4, GJB1, SH3TC2, HK1, GDAP1, SPTLC1, SPTLC2 and MFN2. The duplication of PMP22 responsible for CMT1A was tested by MLPA.
Between 2012 and 2014, 102 patients with IPN were analysed through Sanger sequencing and MLPA. Ninety patients had CMT1 and 12 patients had CMT2. 46/102 (45%) patients had a duplication of PMP22. The remaining 56 patients were analysed by Sanger sequencing of candidate genes. A mutation was found in 13 patients (23% of positive cases): four mutations in MPZ, four mutations in GJB1, two mutations in GDAP1, two mutations in MFN2 and one mutation in HK1. Out of the 44 patients with CMT1, we achieved the molecular diagnosis in 25% of them, while only 2 out of 12 patients with CMT2 (17%) could be ascertained. Among the remaining 43 negative cases, 17 were analysed by targeted analysis of 81 genes from NGS data and for 10 of these 17 patients, we identified the molecular defect in a gene unexplored by Sanger sequencing.
Between 2015 and 2017, a pathogenic variant was found in 49 patients (40%) by targeted analysis of 81 genes from NGS data. During the same period, the PMP22 duplication was identified by MLPA in 67 patients.
In conclusion, in our laboratory, we were able to reach a diagnosis in 23% of patients using Sanger sequencing and in 40% of patients when using targeted NGS of 81 IPN genes. When including PMP22 duplication, 63% of patients with CMT1 were able to get a diagnosis with Sanger analysis against 87% with NGS. In parallel, we were able to make a diagnosis for 17% of CMT2 patients with Sanger analysis against 36% with NGS.
Our comparative statistical analysis allows us to conclude that the molecular diagnosis yield by targeted NGS was two times higher than molecular diagnosis by Sanger sequencing (OR=2.18, p=0.04, (1.0224; 4.8954)).
We compared the solve rate between sporadic cases and family cases. Within our cohort of 123 patients, 42 were sporadic cases; we found a potentially pathogenic variant in 8 of them (19%). Conversely, 51 were familial cases following an autosomal inheritance mode, 25 patients (49%) were able to have a molecular diagnosis.
One of the other objectives of this study was to search for CNVs in our cohort. We found one triplication and one duplication in FGD4 and TFG genes in two patients with CMT1 and CMT2, respectively, as well as a duplication in one of the 81 genes in patient 28 for whom we also found two heterozygous variants in LITAF and DCTN1 using targeted NGS (see table 2B). Overall, ExomeDepth and Q-PCR enabled us to make a potential molecular diagnosis for two additional patients and consequently raise our percentage of diagnosis to 41%.
Discussion
Before targeted NGS, a strategy of sequential molecular diagnosis through Sanger sequencing was implemented. Genes to be screened were chosen according to phenotype, inheritance and electrophysiological criteria. Therefore, the strategy relied mainly on genotype–phenotype correlation. Targeted NGS allows for a more comprehensive analysis with broader panels of genes, faster and cost-effective outcomes.13 14
In our laboratory, we chose targeted NGS in order to avoid the interpretation of numerous variants generated with other NGS strategy, such as whole exome sequencing (WES) or whole genome sequencing, in particular when only one individual is available in the same family.
The use of NGS allowed us to raise our rate of molecular diagnosis to 87% for CMT1% and 36% for CMT2. The high success rate obtained for CMT1 with NGS is due to the high prevalence of PMP22 duplication in this disease subgroup. The PMP22 duplication is responsible for CMT1A, the most frequent CMT subtype, accounting for 48.8% to 63.2% of all subtypes.15–17 Several publications have shown that mutations in GJB1, MFN2, MPZ and PMP22 account for 80 to 94.9% of CMT, and recommended to first complete targeted Sanger sequencing based on clinical phenotype. MFN2 was the most frequently mutated gene found among our cohort of 64 patients with CMT2. NGS enabled us to find a high frequency of pathogenic variants in NEFL, GAN, AARS and KIF5A. These were less frequent and we would not have explored them as first line. Other variants found in BAG3, BICD2, DYNC1H1, REEP1 and FAM134B are much more rare. A survey of 17 880 patients with CMT compared the diagnosis outcomes of Sanger-MLPA and NGS-MLPA17 analyses. This study suggested that the frequency of positive results for 14 CMT genes was not significantly different (p<0.05) in spite of differences in testing strategies between Sanger and NGS. But the bias of this survey is that it compared the same genes in the two groups.
In our cohort of 123 patients, NGS enabled us to make a positive molecular diagnosis of point mutation (single-nucleotide variation (SNV)) for 49 patients (40%) while saving considerable time and cost. This result is consistent with those found in previous studies, including Hartley et al,18 who were able to make a molecular diagnosis by WES in 12 patients (24%) in a cohort of 50 families with IPN. Similarly, Gonzaga-Jauregui et al 4 was able to make a molecular diagnosis in 17 patients (46%) by WES in a cohort of 37 families with CMT.
Recently, Dorhn and collaborators19 describe a cohort of 612 subjects who came from Germany affected by IPN and found a majority of point mutation in MPZ, MFN2, GJB1 and SH3TC2. Our study describes patients who came from the south of France and found a majority of mutation in MFN2, SH3TC2, NEFL, GAN, GDAP1, AARS and KIF5A. In fact, our cohort is composed of people who came from the Mediterranean region which probably explains the different spectrum delineation of our study, notably the relatively high frequency of GAN variants.
Moreover, 11 patients in our cohort were undiagnosed but we found a potentially pathogenic variant without confirmation of pathogenicity. In the following, we will detail some cases of particular interest.
Boyer and collaborators20 have described nine mutations in INF2 in 12 patients with CMT with focal segmental glomerulosclerosis. In our study, patients 5, 27, 35, 45 and 52 have a sequence variation in this gene and, to date, no nephrological disease is known in these patients. Patients 5 and 45 have a probably pathogenic variant according to Richards classification. Those mutations are located in the same codon that patient described by Boyer. While patient 27, 35 and 52 have a VUS according to Richards classification. At that time, without nephrological examination and familial study, we were not able to exclude those variants.
We have two patients in this cohort affected by CMT1 (patient 21) and CMT2 (patient 38) carrying numerous variants in potential genes. We found three VUS in TRPV4 (CMT2C), LRSAM1 (CMT2P) and KIF1B (CMT2A1) genes for patient 21. Mutations in these three genes were reported to be in relation with CMT type 2 or dHMN. In that case, we have too much variant which could explain patient’s phenotype at isolated state or in association. A familial study is indicated. Patient 38 showed two VUS in SEPT9 (hereditary neuralgic amyotrophy)21 and ARHGEF10 (slowed nerve conduction velocity).22 These mutations could thus separately or in association explain these patient’s phenotype. Unfortunately, the family study could not be achieved and we could not make any conclusions related to the contribution of these variants in the disease.
Only one variant was classified as probably benign in Richards classification, and namely variant c.830G>A in GJB1 which was identified in patient 16 suffering from autosomal-dominant CMT1 with first clinical manifestations at the age of 15. She presents a severe phenotype with standard clinical signs of peripheral motor and sensory neuropathy. Motor nerve conduction velocity was 36 m/s in the electromyogram and the nerve biopsy showed Schwann cell proliferation in the form of an onion bulb. The segregation analysis found this variant in the asymptomatic mother. Her mother could have an inactivation of the mutant allele on one of the X-chromosomes, thus only expresses wild-type Cx32 from the normal allele. NGS analysis did not enable us to identify another potential variant responsible for her neuropathy. Patient 4, affected with autosomal-dominant CMT2, showed severe clinical signs at the age of 9. We found a VUS c.67C>G, (p.Arg23Gly) in NEFL (CMT1F, CMT2E). The segregation analysis found this one in the asymptomatic mother. This case can maybe illustrate incomplete penetrance for this mutation in this family. Alternatively, the disease, in this patient, is due to another mutation in a gene not explored in this targeted NGS.
Patients 13 and 56 are affected with HSAN and carry a mutation in SPTLC2, known to be responsible for HSAN1C (MIM 613640). They also have a VUS (according to Richards classification) in another gene. One in HSPB3 (HMN2C, MIM 613376) for patient 13 and the second one in MYH14 (peripheral neuropathy, myopathy, hoarseness and hearing loss, MIM 614369) for patient 56. These two patients have perforating ulcers of the foot, hypoaesthesia at sock level but their motor picture also included a severe progressive motor deficiency, severe wasting, contractures and neuropathic pain. In these cases, SPTLC2 could alone be responsible for the patient’s phenotype and we can consider HSPB3 and MYH14 as modifiers or modulators factors likely to account for the motor phenotype of these two patients. In comparison, Sinkiewicz-Darol’s team23 showed that the presence of a variant p.Ile92Val in gene LITAF/SIMPLE of patients who presented with a duplication or deletion of PMP22 was linked with an earlier onset of CMT1A or Hereditary Neuropathy by Hypersensibility to Pressure Palsie (HNPP) and could be considered as a modifier. These additional variants could contribute to the variability of the clinical phenotype expression.
Even if the majority of IPNs are explained by SNV, CNV may equally be a genetic cause of IPN and, thanks to NGS strategy, we can now detect these two types of variations. We found three CNVs in three patients, that is, 2.4% in our cohort. In comparison, Pfundt’s team24 looked for CNVs with the coNIFER software using reading depth data with exome high-throughput sequencing in a cohort of 2603 patients who had different genetic pathologies. In the group with neurodevelopment disorders, muscular disorders and abnormal coordination, they respectively found 1.3%, 0.6% and 0.9% of CNV. We found a higher percentage of CNVs in our IPN group (none significant Fisher’s exact test). This study of CNV enabled us to raise our rate of molecular diagnosis to 51 patients (41%) out of a cohort of 123 patients.
Targeted NGS allows us to improve our molecular diagnosis in IPN and allows an accurate genetic counselling in families. Moreover, a positive molecular diagnosis is the first step to participate in clinical trials.
Conclusion
In our laboratory, NGS improved the molecular diagnostic rate, allowing for 40% of the patients suffering from IPN. Eighty-seven per cent of patients with CMT1 can now get a precise molecular diagnosis. On the other hand, 64% of CMT2 cases remain unsolved. Indeed, numerous aetiologies of neuropathies are found in elderly patients, such as diabetic, inflammatory, alcoholic, idiopathic and autoimmune neuropathies and may be confused with hereditary neuropathies. Most importantly, NGS analysis allows to describe novel mutations, not yet reported in the literature, that are of significant importance for the scientific and medical community. Moreover, the analysis of NGS data enabled us to detect possible duplications and deletions of genes not investigated routinely. Targeted NGS with a panel of 81 genes is therefore well adapted to IPN molecular diagnosis. However, the generation of many variants of unknown significance requires a collegial interpretation by biologists, geneticists and neurologists.
Acknowledgments
The authors would like to thank the patients and their families for their kind cooperation. They also thank Caroline Lacoste and Christophe Pécheux for supplying the NGS platform and Catherine Badens for helping.
References
Footnotes
Contributors JB, NB-P, VD and NL contributed to the design of the study. NB-P contributed to the supervision and mentorship. JB, AB, J-PD and NM participated in data analysis and interpretation. JB, TS and NB-P contributed to manuscript drafting. TS, FA, BC, ES-C, ED, AV, SA, AC and NL contributed to the collection of clinical data during consultations. All authors participated in critical revisions of the manuscript and can take responsibility for its integrity and the accuracy of the data analysis.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent obtained.
Ethics approval Aix-Marseille University Ethics Comittee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The data are not available freely. Enquiries and requests for further information should be made to corresponding author.