Article Text

Abstract
Introduction Oral corticosteroids are the first-line treatment for idiopathic childhood nephrotic syndrome. Most children experience several relapses, needing repeated courses of corticosteroid therapy. This exposes them to side effects and long-term complications. For most patients, long-term prognosis is for complete resolution of the disease over time and maintenance of normal kidney function. Therefore, it is vital to focus on minimising adverse events of the disease and its therapy. Unfortunately, no randomised controlled trials are available to determine the optimal corticosteroid treatment of an infrequent relapse of nephrotic syndrome. Recent studies show that treatment schedules for the first episode can safely be shortened to 2 months. The hypothesis of the REducing STEroids in Relapsing Nephrotic syndrome (RESTERN) study is that a 4-week reduction of alternate-day steroids after inducing remission is effective and safe, reduces steroid exposure by 35% on average and is therefore preferable.
Methods and analysis The RESTERN study is a nationwide, double-blind, randomised, placebo-controlled, non-inferiority intervention study. Children aged 1–18 years with a relapse of steroid-sensitive nephrotic syndrome are eligible for this study. Study subjects (n=144) will be randomly assigned to either current standard therapy in the Netherlands or a reduced prednisolone schedule. The primary outcome of the RESTERN study is the time to first relapse after the final prednisolone dose. The secondary outcomes are the number or relapses, progression to frequent relapsing or steroid dependent nephrotic syndrome and the cumulative dosage of prednisolone during the study period.
Ethics and dissemination This non-inferiority trial will be performed in accordance with the Declaration of Helsinki and has been approved by the medical ethical committee of Arnhem-Nijmegen and the Dutch Competent Authority (Central Committee on Research Involving Human Subjects, CCMO). After completion of this study, results will be published in national and international peer-reviewed scientific journals. Papers will be published according to CCMO guidelines. The final report will be made available to trial participants.
Trial registration number NTR5670, EudraCT no 2016-002430-76.
- nephrotic syndrome
- pediatrics
- corticosteroids
- randomised clinical trial
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Double-blind, randomised, placebo-controlled study.
Nationwide inclusion.
Large study cohort.
Two common practices in the Netherlands regarding the current treatment of relapsing nephrotic syndrome.
Side effects and toxicity of steroids might jeopardise the double-blind design of the study.
Introduction
Nephrotic syndrome is characterised by the triad of severe proteinuria, hypoalbuminaemia and oedema. It is one of the most common glomerular diseases in children with an incidence of 1–7 per 100 000 children per year (Dutch data: 1.52/100 000) and a prevalence of 16 per 100 000 children.1–3 Most children have minimal change nephrotic syndrome and will have favourable prognosis with complete resolution of the disease over time and maintenance of normal kidney function.4
For over 60 years, corticosteroids have been the first-line treatment for idiopathic nephrotic syndrome in children as over 80%–90% of patients achieve complete remission after prednisolone treatment.5 6 Yet, over 80% of the patients experience one or more relapses and around 50% suffer from frequent relapses, thereby needing additional courses of corticosteroid therapy.7 This exposes them to the side effects and long-term complications of corticosteroid therapy, such as growth retardation (8%–16%), osteopaenia (13%–63%), mood disorders and cataract (6%–20%).8–11 The currently used treatment regimens for a nephrotic syndrome relapse are mostly based on practice guidelines of the International Study of Kidney Disease in Children (ISKDC)12 and the Arbeitsgemeinschaft für Pädiatrische Nephrologie (APN).13 In the Netherlands, the standard treatment schedule consists of 60 mg/m2 prednisolone daily until complete remission is achieved for 3 days, followed by 40 mg/m2 prednisolone on alternate days for 4–6 weeks.14 15
Several trials have been conducted to study the duration of the initial corticosteroid treatment regimen, as Hodson et al suggested that a prolonged period of prednisolone might reduce chances of subsequent relapses.16 A previous nationwide study in the Netherlands addressed the issue of duration of corticosteroids for the initial presentation and showed that the duration had no impact on subsequent relapses.7 A few recent, well-conducted trials suggest that it may be safe to reduce the duration and thereby cumulative dose of corticosteroid therapy for the initial episode of nephrotic syndrome from 6 to 2 or 3 months.17–21 With recent studies showing no benefit for longer duration of initial prednisolone treatment, one may conclude that we still don’t know the optimal treatment duration of relapses. In addition, as stated in the KDIGO (Kidney Disease: Improving Global Outcomes) clinical practice guideline glomerulonephritis, “there are no RCTs examining relapse regimens with corticosteroids in infrequent relapsing nephrotic syndrome”.22 With the current evidence against longer steroid therapy for the initial treatment, the time is now to determine whether this holds true for treatment of relapses as well, in both children with and without maintenance immunosuppressive therapy.
The aim of the RESTERN study (REducing STEroids in Relapsing Nephrotic syndrome) is to assess the safety and effectiveness of a reduced alternate day steroid schedule for treatment of a nephrotic syndrome relapse in comparison with the current standard therapy.
Methods and analysis
Trial design and setting
The RESTERN study is designed as a nationwide, double-blind, randomised, placebo-controlled, non-inferiority intervention study with two treatment arms. The study is performed and coordinated by a single centre (Radboudumc Amalia Children’s Hospital) where the research team is instituted, with inclusion of patients throughout the Netherlands from all secondary and tertiary hospitals.
Eligibility criteria
Children aged over 1 and less than 18 years with steroid-sensitive nephrotic syndrome will be assessed for possible inclusion in the study. A detailed description of the inclusion and exclusion criteria is shown in box.
Inclusion and exclusion criteria
Inclusion criteria
Age over 1 and less than 18 years
Steroid-sensitive nephrotic syndrome
At least one episode of nephrotic syndrome in the preceding 24 months that was treated with prednisolone
The last prednisolone use (at a dose over 10 mg/m2 on alternate days) for the treatment of a previous episode was at least 4 weeks ago
Subjects without maintenance immunosuppressive therapy
Subjects with maintenance immunosuppressive therapy
Long-term immunosuppressive therapies: levamisole, ciclosporin, tacrolimus, mycophenolate mofetil (Cellcept), mycophenolate sodium (Myfortic), prednisolone maximum of 4 mg/m2 on alternate days
Cyclophosphamide (oral or intravenous), at least 3 months postcompletion of therapy
A single dose or course of intravenous rituximab, at least 3 months postcompletion of therapy
Signed informed consent from the parent or legal representative and/or the patient, depending on the age of the patient
Exclusion criteria
Steroid resistant nephrotic syndrome
Receiving or within 3 months after receiving, cyclophosphamide or rituximab
Daily prednisolone maintenance therapy at any dose
Alternate-day prednisolone maintenance therapy at a dose over 4 mg/m2
Documented or suspected significant non-compliance
Pregnancy
Stimulant drug use
Comorbidity
Kidney transplant recipient
Any disease that requires the variation in oral prednisolone to be at the discretion of the treating physician(s)
Concomitant use of moderate and strong CYP3A inducers
Concomitant use of moderate and strong CYP3A inhibitors, other than ciclosporin
Study objectives
The primary objective of this study is to investigate the effectiveness of a reduced steroid schedule for the treatment of a relapse in children with steroid-sensitive nephrotic syndrome. Secondary objectives are the following:
To study the influence of maintenance immunosuppressive therapy on the effectiveness of a reduced steroid schedule for the treatment of a relapse in children with steroid-sensitive nephrotic syndrome. Maintenance immunosuppressive therapies include levamisole, ciclosporin, tacrolimus, mycophenolate mofetil and mycophenolate sodium, and alternate-day prednisolone with a maximum of 4 mg/m2;
To investigate the occurrence of relapses, frequency of relapses and progression to steroid dependent and frequent relapsing nephrotic syndrome in children with nephrotic syndrome under the standard treatment regimen;
To study the influence of maintenance immunosuppressive therapy on the occurrence of subsequent relapses, frequency of subsequent relapses and progression to steroid dependent and frequent relapsing nephrotic syndrome under the standard regimen;
To study the effectiveness of a reduced steroid schedule for the treatment of a relapse and occurrence and frequency of subsequent relapses in children with steroid dependent nephrotic syndrome.
Interventions
Eligible patients will be randomised between standard prednisolone treatment and a reduced treatment schedule. At the start of a relapse, participants are treated according to the current standard therapy, consisting of daily oral prednisolone (60 mg/m2). After 3 days of remission, defined as 3 consecutive days of absent proteinuria based on urine dipstick analysis, standard care dictates that prednisolone is changed to an alternate-day dosing of 40 mg/m2 with a maximum of 40 mg. After 2 weeks of alternate-day prednisolone, participants are randomised between the two treatment arms. The standard treatment group will receive an additional 4 weeks of alternate-day oral prednisolone (40 mg/m2, with a maximum of 40 mg) and the placebo group will receive 4 weeks of alternate-day oral placebo (figure 1). Prednisolone (5 mg/mL) or placebo will be provided as an oral solution (see Investigational medicinal product section).
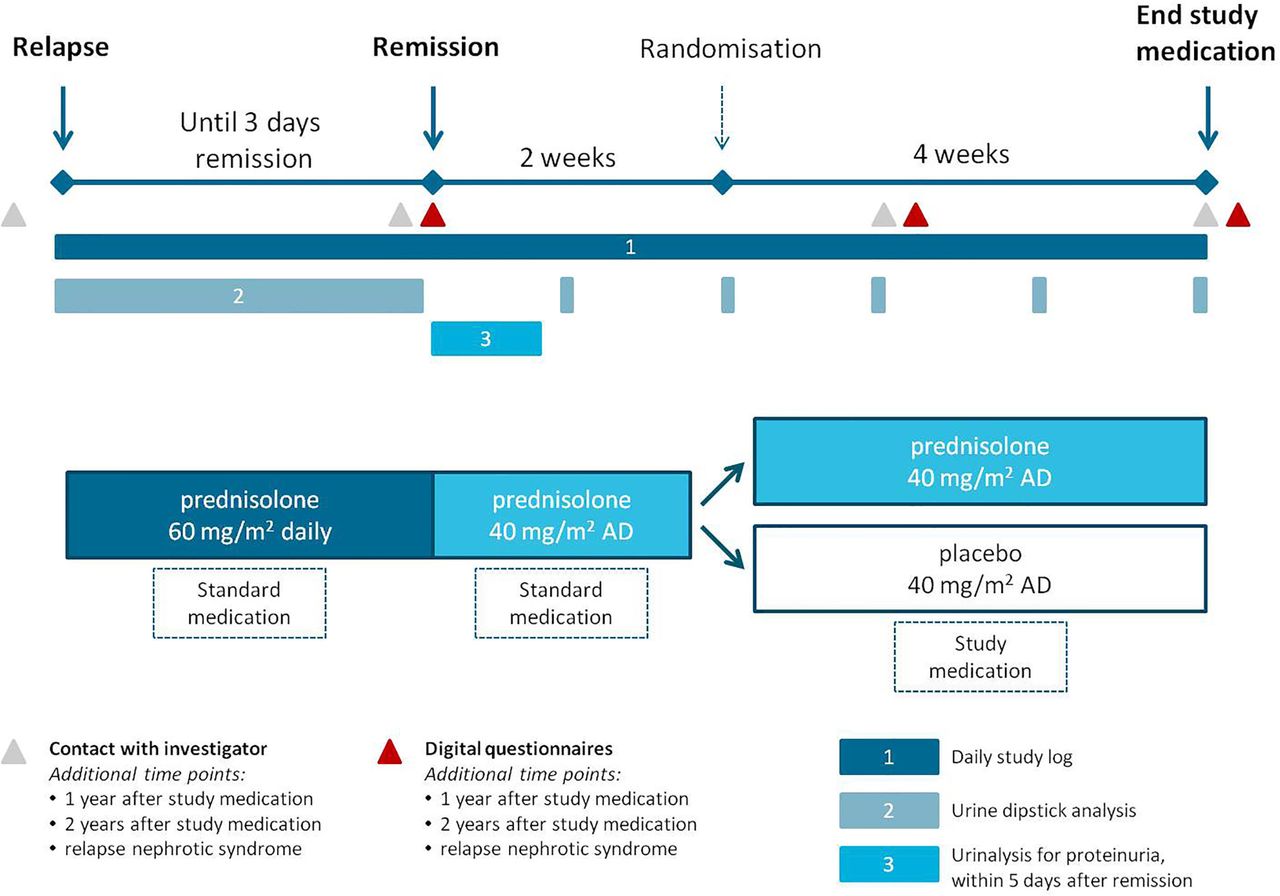
{kind=link}
Intervention schedule and study procedures. AD, alternate days.
Children will be withdrawn if they are unable to take the study medication and will be treated according to the standard treatment regimen (oral prednisolone 40 mg/m2 on alternate days). Maintenance immunosuppressive therapy, including levamisole, ciclosporin, tacrolimus, mycophenolate mofetil and mycophenolate sodium, is continued throughout the treatment period. Alternate-day prednisolone maintenance therapy with a maximum of 4 mg/m2, is discontinued during the non-randomised treatment and restarted after randomisation, administered at the same day as the study medication. Antihypertensive agents, antiproteinuric agents and/or diuretics may be continued at the discretion of the treating physician.
All children will be followed for 2 years and subsequent nephrotic syndrome relapses will be treated according to the current standard treatment protocol in the Netherlands.
Investigational medicinal product
A prednisolone or placebo solution (5 mg/mL) will be produced compliant with current Good Manufacturing Practice at the department of pharmacy of our institute. The standardised formulation of the oral solution is based on the Dutch Pharmacists Formulary. The investigational medicinal product is an aqueous solution preserved with methylparaben, buffered at a pH of 7.1 with a phosphate buffer and contains sorbitol and banana essence to mask the bitter taste of prednisolone sodium phosphate. For the placebo, prednisolone sodium phosphate is left out of the product. A pilot palatability study of the investigational medicinal product showed no relevant visual or taste differences of the drug or placebo. Drug dispensing and accountability is performed on individual basis from the central pharmacy.
Outcomes
The primary outcome of the RESTERN study is the time to first relapse. This is defined as the time (in days) from the final prednisolone dose until the first day of treatment of the next relapse.
Secondary outcomes include the following:
The number of relapses per patient after the final prednisolone dose, censored at 12 and 24 months of follow-up;
Progression to frequent relapsing nephrotic syndrome, defined as four or more relapses in any 12-month period (KDIGO criteria), censored at 24 months of follow-up;
Progression to steroid dependent nephrotic syndrome, defined as two consecutive relapses during corticosteroid therapy or within 14 days of ceasing therapy (KDIGO criteria), censored at 24 months of follow-up;
Cumulative dosage of prednisolone (mg/m2) during study period, censored at 12 and 24 months of follow-up.
Participant timeline
During the period of daily prednisolone, participants determine the timing of remission by daily urine dipstick analysis. In order to objectively establish remission of nephrotic syndrome, participants are requested to deliver a urine sample to the local hospital within 5 days of attaining remission to confirm the absence of proteinuria. During the 2 weeks of alternate-day prednisolone and the subsequent 4 weeks of study medication, participants are requested to check their urine for proteinuria at least weekly. In addition, patients are requested to fill out digital questionnaires at different time points (figure 1). As shown in table 1, follow-up information will be collected at 1 and 2 years after randomisation and when a relapse occurs.
Study questionnaires
Sample size
The sample size calculation is based on the non-inferiority design and calculated for the primary outcome: time to first relapse after the final prednisolone dose. Based on previous data, average time to relapse in the first year is approximately 185 days with an SD of 120 days.7 Using the power calculation for a non-inferiority trial with a continuous primary outcome, a power of 80% and a non-inferiority limit of 50 days, 72 patients per group are required. Using a Cox proportional hazard time-to-relapse analysis (survival analysis), similar numbers can be calculated. With an estimated prevalence of nephrotic syndrome of 15 in every 100 000 children, a population most at risk between the ages of 2 and 12 years, about 270 children may be at risk of developing a nephrotic syndrome relapse each year. The necessary inclusion rate is therefore approximately 50%. Subjects will be replaced after withdrawal. Based on withdrawal rates of a previous nephrotic syndrome clinical trial in the Netherlands7 a maximum of 23 subjects (16%) will be replaced. The reason for withdrawal will be recorded in the medical status report and the trial master file.
Recruitment
Study subjects will be notified about the existence of the RESTERN study via their treating physician, the patient associations and/or the study website (www.restern.nl). Written informed consent for participation will be obtained from the parents or legal representative(s) and/or the patient, depending on the age of the patient.
Randomisation and blinding
Participants will be randomly allocated in a 1:1 ratio to receive either prednisolone or placebo. The randomisation will be performed by the pharmacy of our institute using the data management system Castor Electronic Data Capture23 with stratification for treatment with immunosuppressive maintenance therapy. Castor uses a variable block algorithm with random blocks of 4, 6 or 8. The randomisation list remains preserved by the pharmacy and will not be accessible to the investigators until the end of the follow-up of the last patient. An unblinding procedure at the hospital pharmacy department will be available at all times. The true group allocation will be unmasked only if necessary and after the database is locked.
Data collection
Participants are requested to maintain a digital study log in which results from dipstick analysis, medication and special remarks are gathered. In addition, participants receive digital questionnaires about their medical history, previous relapses and the current nephrotic syndrome relapse. Moreover, the digital questionnaires include questions about side effects and steroid toxicity at different time points, for example, during the period of daily prednisolone, alternate-day prednisolone and study medication. First, patients are asked if the different side effects were present during the specific time period. Second, the level of inconvenience is assessed (ranging from not at all inconvenient to very inconvenient). Local paediatricians and paediatric nephrologists will be requested to provide patient information at different time points. Patients randomised who did not take their allocated treatment will be considered as having deviated from the protocol. If a patient or their representative withdraws consent for data collection, only data up to the point of withdrawal will be used in the analysis.
Data management
The study will use the Good Clinical Practice (GCP) compliant, web-based application Castor Electronic Data Capture to record data.23 Data will be entered in the case report form in Castor by the coordinating investigators at the Radboudumc. The digital questionnaires will automatically be uploaded in the data management system.
Statistical analysis
Statistical analysis will be conducted using IBM SPSS Statistics. A p value <0.05 will be considered statistically significant. The main analysis will consist of an intention-to-treat analysis. Participants who are lost to follow-up or in whom trial medication is stopped prematurely will be analysed according to their allocated groups. In addition, as intention-to-treat analysis may increase the risk of type 1 errors in a non-inferiority trial, a per-protocol analysis will also be conducted.24 Missing baseline and outcome data will not be imputed. When a patient is lost to follow-up or has withdrawn consent, all available data up until withdrawal of consent or loss to follow-up will be used. Discrete variables will be summarised by frequencies and percentages. Continuously distributed variables will be summarised using either mean and SD for data with normal distribution or median and IQR for non-normally distributed data. Further details regarding statistical analysis of the primary and secondary outcomes can be found in the statistical analysis plan (online supplementary file 1).
Supplementary file 1
Monitoring
As the standard treatment group will receive the current standard therapy for a nephrotic syndrome relapse in the Netherlands, no specific safety surveillance is needed for this group. The placebo arm provides the participants with a reduced prednisolone exposure, which is therefore unlikely to result in any adverse events. However, the main concern in this study is that the reduced treatment schedule may result in an earlier relapse, which is the primary endpoint of the study. An external Data Safety and Monitoring Board (DSMB) will be convened to monitor safety outcomes and to provide the principal investigator with recommendations regarding reconsideration of the trial. The DSMB will consist of two members: a methodologist and a paediatric nephrologist with experience in clinical trials, both independent of the trial. Interim analysis performed by the DSMB will take place 3 months after the first 40 participants have received study medication. Aim is to check for a significant difference in relapse rate between the two groups. Further details about the interim analysis can be found in the statistical analysis plan. An independent research coordinator will monitor the study to verify that the rights and well-being of human subjects are protected, the reported trial data are accurate, complete and verifiable from source documents and the conduct of the trial is in compliance with the currently approved protocol and GCP. The coordinating investigators will report the serious adverse events and will submit an annual safety report to the medical ethical committee and competent authority.
Ethics and dissemination
Ethics approval
The RESTERN study has been approved by the medical ethical committee of Arnhem-Nijmegen and the Dutch Competent Authority (Central Committee on Research Involving Human Subjects, CCMO). The registration number of the RESTERN study is NL8185.091.16. The project will be conducted in line with the Declaration of Helsinki. In addition, all researchers will follow the guidelines for GCP and trial outcomes will be reported in line with the Consolidated Standards of Reporting Trials guidelines.25 Any substantial amendments or modifications of the protocol will be presented to the medical ethical committee and, when approved, be notified to the Competent Authority compliant with European regulations.
Consent
Written informed consent for participation will be obtained from the parents or legal representatives and/or the patient, depending on the age of the patient. Patients will be informed that withdrawal from the study is possible at any time at their own discretion without necessarily giving reasons. The ‘code of conduct involving minors’ will be used as guideline to respond appropriately to resistance of subjects to study procedures as established by the Paediatric Association of the Netherlands.
Confidentiality
All patients have their own unique patient identification number as allocated by the hospital administration. Source data will be stored confidentially in the hospital information system under the subject’s identification number. Participants will also receive an identification code, all final study data will be kept under this identification number. The investigators safeguard the key to the code. Handling of personal data will comply with the Dutch Personal Data Protection Act. Data will be stored until 15 years after publication.
Dissemination policy
The trial is registered on the Dutch Trial Registry, trial number NTR5670, prior to the start of inclusion.26 After completion of this study, results will be published in national and international peer-reviewed scientific journals. Papers will be published according to CCMO guidelines. The final report will be made available to trial participants.
Discussion
The RESTERN study aims to demonstrate that relapses of nephrotic syndrome in children can be treated effectively and safely by a reduced duration of alternate day prednisolone. Using a nationwide, double-blind, randomised, placebo-controlled, non-inferiority study, the hypothesis will be tested.
Currently, corticosteroid treatment duration in children with infrequent relapses of steroid-sensitive nephrotic syndrome is based on empirical recommendations from the ISKDC and APN. The RESTERN study is the first randomised placebo-controlled clinical trial to investigate a reduced corticosteroid schedule for the treatment of relapsing nephrotic syndrome in childhood. So far, most studies have been conducted to investigate the initial treatment schedule. Recently, it has been shown that a reduction in prednisolone duration for the treatment of a first presentation of nephrotic syndrome in children, with or without increased cumulative dosage, is clinically safe and results in similar treatment outcomes, while potentially reducing side effects.17 18 20 21 For frequent relapsing nephrotic syndrome, an abstract from a single randomised controlled trial suggests that children with relapsing steroid-sensitive nephrotic syndrome relapse less frequently if treated with tapering doses of prednisolone for 7 months compared with the standard treatment of 2 months. Unfortunately, these results have never been published, which makes it impossible to examine them closely and evaluate for any bias.18 27
In this study, a treatment duration of 6 weeks alternate-day prednisolone after inducing remission for the standard therapy group was chosen as this is the current standard therapy in the Netherlands for the treatment of a nephrotic syndrome relapse. As the KDIGO recommendation of at least 4 weeks alternate-day steroid treatment is based on a rather small study,28 the Dutch guidelines traditionally follow the 6-week alternate-day steroid schedule described by the APN. However, a potential limitation of this study could be that some clinicians already reduced the alternate-day treatment schedule from 6 to 4 weeks after inducing remission based on the notion of this in the KDIGO guideline.22 Our choice of 6 weeks alternate-day prednisolone may therefore discourage eligible patients to participate in our study as this may increase the prednisolone duration for some patients. The use of different treatment protocols in the Netherlands underlines the need for our randomised controlled trial to determine the optimal dosing regimen for a nephrotic syndrome relapse. In case non-inferiority is shown, our results are also transferable to the KDIGO recommendation of at least 4 weeks of alternate-day steroid treatment. However, in case inferiority is shown, additional research is needed.
The results of the RESTERN study may provide evidence to adapt current recommendations for national and possibly international guidelines to treat children with relapsing nephrotic syndrome. If corticosteroid exposure could be reduced to treat relapses of nephrotic syndrome, this would reduce the toxicity of prednisolone and thereby decrease the side effects and long-term complications associated with corticosteroid therapy in children with relapsing nephrotic syndrome.
Trial status
The study started recruitment in December 2016 and is currently recruiting.
References
Footnotes
Contributors MFS is the principal investigator of the RESTERN study, initiated the project and drafted the protocol. AMS drafted this manuscript based on the METC approved protocol using the SPIRIT checklist. All authors critically reviewed and revised the manuscript and approved the final manuscript as submitted.
Funding This work was supported by a Senior Postdoc Grant to MFS from the Dutch Kidney Foundation, project number 15OKG16.
Competing interests None declared.
Ethics approval The RESTERN study has been approved by the medical ethical committee of Arnhem-Nijmegen and the Dutch Competent Authority (Central Committee on Research Involving Human Subjects, CCMO). The registration number of the RESTERN study is NL8185.091.16, file no 2016-2288.
Provenance and peer review Not commissioned; externally peer reviewed.