Article Text

Abstract
Introduction There is a need to find alternatives to the use of human donor corneas in transplants because of the limited availability of donor organs, the incidence of graft complications, as well as the inability to successfully perform corneal transplant in patients presenting limbal deficiency, neo-vascularized or thin corneas, etc. We have designed a clinical trial to test a nanostructured fibrin-agarose corneal substitute combining allogeneic cells that mimics the anterior human native cornea in terms of optical, mechanical and biological behaviour.
Methods and analysis This is a phase I-II, randomised, controlled, open-label clinical trial, currently ongoing in ten Spanish hospitals, to evaluate the safety and feasibility, as well as clinical efficacy evidence, of this bioengineered human corneal substitute in adults with severe trophic corneal ulcers refractory to conventional treatment, or with sequelae of previous ulcers. In the initial phase of the trial (n=5), patients were sequentially recruited, with a safety period of 45 days, receiving the bioengineered corneal graft. In the second phase of the trial (currently ongoing), subjects are block randomised (2:1) to receive either the corneal graft (n=10), or amniotic membrane (n=5), as the control treatment. Adverse events, implant status, infection signs and induced neovascularization are evaluated as determinants of safety and feasibility of the bioengineered graft (main outcomes). Study endpoints are measured along a follow-up period of 24 months, including 27 post-implant assessment visits according to a decreasing frequency. Intention to treat, and per protocol, and safety analysis will be performed.
Ethics and dissemination The trial protocol received written approval by the corresponding Ethics Committee and the Spanish Regulatory Authority and is currently recruiting subjects. On completion of the trial, manuscripts with the results of phases I and II of the study will be published in a peer-reviewed journal.
Trial registration CT.gov identifier: NCT01765244 (Jan2013). EudraCT number: 2010-024290-40 (Dec2012).
- corneal transplantation
- corneal ulcer
- stromal fibrosis
- limbal stem cell deficiency
- tissue bioengineering
- randomized controlled trial
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- corneal transplantation
- corneal ulcer
- stromal fibrosis
- limbal stem cell deficiency
- tissue bioengineering
- randomized controlled trial
Strengths and limitations of the study
This is a prospective, phase I-II, randomised, open-label clinical trial that will evaluate the safety and feasibility, as well as clinical efficacy evidence, of a bioengineered corneal substitute in adults with severe trophic corneal ulcers. This model of human anterior allogeneic cornea will provide an alternative approach in cases where human donor keratoplasty is not an option.
To our knowledge, this is the first report showing an investigator-driven multicenter randomised clinical trial evaluating bioengineered anterior corneal implants combining two types of allogeneic cells (epithelial cells and corneal fibroblasts). The design, management and coordination efforts invested on this trial may be of special interest to serve as a model for emerging multicenter investigator-driven, publicly funded clinical trials on tissue engineered products.
This study has been designed as a small size randomised controlled trial due to limitations in the expiration period of the investigational product, which considerably restricted site selection process. Wide eligibility criteria allow for an easier screening and recruitment, making the study more feasible and economical. Although this strategy may provide a heterogeneous sample, we believe it will not interfere with the comparison of the main safety outcome, as we are measuring prognostic variables at baseline and throughout follow up to allow for a precise discrimination of causality of the events.
The special characteristics of the trial interventions impeded the possibility of blinding, which is a limitation to the study design. To avoid evaluation bias both allocation groups follow-up procedures are equivalent, standardized grading systems are used when possible for objective outcome measurements and statistical analysis will be performed by blinded data analysts.
Introduction
Corneal blindness is the third leading cause of blindness worldwide, after cataract and glaucoma.1 2 Bilateral corneal blindness is estimated to occur in 4,9 million individuals globally, with 23 million people suffering from unilateral corneal blindness.3 Moreover, corneal diseases cause 20% of the prevalence of childhood blindness.4 Currently, the primary treatment for corneal blindness is donor corneal transplantation.2 However, there is a severe shortage of good quality corneas worldwide,5 and the problems arising from keratoplasty (waiting lists, complications and graft failure, slow graft integration, quality control of donor tissue, etc.),6 7 as well as the inability to successfully perform the transplant in some groups of patients (cases with limbal stem cell deficiency, neo-vascularized corneas, thin corneas, acute corneal burns, etc) make it necessary to develop complementary solutions based on corneal bioengineering.7
Tissue engineering has been applied in ophthalmology aiming to develop an optimal human corneal substitute for more than 20 years. However, only one research group has come close to restitute the damaged corneal stroma of several patients by implanting an acellular collagen-based scaffold in a clinical trial.8–10 After 4 years of follow-up, the scaffold was innervated and repopulated with corneal cells from the patient. Nevertheless, those patients did not present limbal stem cell deficiency (LSCD) or corneal neovascularization. Corneal diseases with damaged stroma associated with LSCD require not only replacing the corneal stroma but also repopulating with limbal epithelial stem cells (LESC).
Different approaches have been proposed to provide LESC in patients suffering LSCD. The most recent techniques used are: transplant of cultured LESC using different substrates (ie, fibrin, amnion, etc),11 and simple limbal epithelial transplantation, where small pieces of limbus are placed directly on the corneal surface using amnion and fibrin glue.12 When the LSCD associates a damage of the corneal stroma or the endothelium, a corneal transplant is necessary to be performed in conjunction with those techniques,11 unless a keratoprosthesis is implanted instead.13 In this context, the generation of an artificial corneal substitute, mechanically and optically suitable, where corneal stem cells could be cultured and transplanted, could emerge as an ideal therapeutic approach for those patients.
In this regard, we developed a tissue-engineered scaffold based on a mixture of fibrin and 0.1% agarose, where corneal epithelial cells, corneal fibroblasts and corneal endothelial cells were co-cultured.14 15 Moreover, the human corneal epithelial cells co-cultured on top of the fibrin-agarose scaffold with human corneal fibroblast cultured inside of the biomaterial, promotes corneal differentiation of the epithelium and proper corneal differentiation of the artificial tissue.15 Furthermore, we demonstrated that these co-cultured fibrin-agarose scaffolds present optimal mechanical and optical behaviour, similar to a native human cornea, after applying a plastic compression process or nanostructuration.16 17 Finally, the high biocompatibility of fibrin-agarose scaffolds was demonstrated in several in vivo models where no signs of rejection or inflammation were found after its implantation in oral mucosa,18 skin19 and peripheral nerve.20
After demonstrating that this model of human anterior allogeneic nanostructured lamellar cornea could mimic the human native anterior cornea in terms of optical,17 21 mechanical,16 and biological behaviour,15 we moved forward to the clinical phase. For that purpose, we designed the present phase I - II, randomised, controlled, clinical trial to evaluate its safety and feasibility, as well as evidence of efficacy data in patients suffering from severe trophic corneal ulcers refractory to conventional treatments or with sequelae of previous ulcers. Corneal ulcers refractory to conventional treatments leave persistent corneal epithelial defects that can lead to excessive stromal degradation. Disease progression may lead to corneal thinning or melting, and eventually, corneal perforation. These disorders are the most serious manifestations of neurotrophic keratopathy and other trophic corneal diseases that require aggressive treatments to prevent loss of visual function and even the eye loss. While the clinical diagnosis is readily oriented from the medical history and clinical findings, treatment of these conditions is one of the most difficult and challenging among all corneal diseases due to the lack of etiologic treatment.22–24 Current treatments try to control the symptoms of the patient by improving the lubrication status of the ocular surface (artificial tears, contact lens, punctal occlusion, etc) or by applying part of the missing trophic factors, such us epithelial growth factor or nerve growth factor, that are present in the blood serum.23 25 When conventional treatments fail, corneal melting and perforation can occur in the most advanced stages. In these cases, more aggressive procedures are necessary, such as cyanoacrylate glue, anterior lamellar keratoplasty, conjunctival graft or amniotic membrane (AM) transplants to preserve the anatomical integrity of the cornea.26 27 We hypothesise that our model of human allogeneic nanostructured lamellar anterior cornea can be safely implanted in these cases, showing at least partial efficacy to reconstruct the corneal ulcer or its sequelae. The bioengineered cornea will provide structural elements (the fibrin-agarose scaffold), cellular content (epithelial cells and keratocytes) and growth factors (mainly those provided by the human plasma that is one of the main compounds used to generate the fibrin-agarose scaffold) to regenerate the damaged corneal tissue. To test this hypothesis, the clinical trial that we describe in this report was designed and conducted.
Methods and analysis
Study design
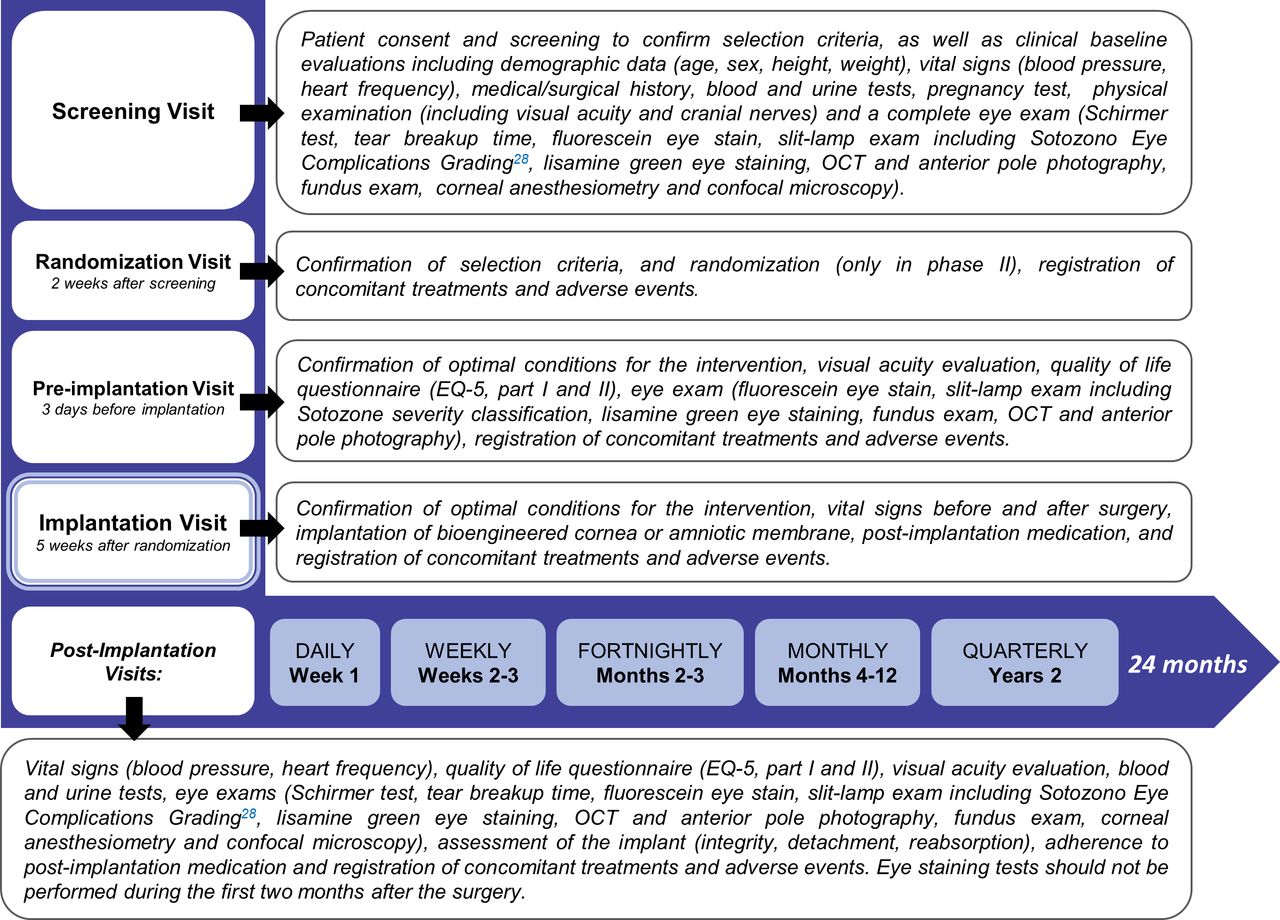
This is a phase I - II, randomised, controlled, open label, multicenter clinical trial, assessing safety and feasibility, as well as evidence of clinical efficacy of a bioengineered human allogeneic nanostructured lamellar anterior cornea, in comparison with AM transplant. Initial phase of the trial comprised sequential recruitment of 5 eligible subjects with a safety period of at least 45 days between each other, receiving the bioengineered corneal graft (non randomised allocation). On completion of a 3 month post-implantation follow-up period, safety and feasibility data were analysed according to the trial’s stopping rules (interim analysis), to allow for continuation of phase II of the study. The remaining fifteen patients that complete the study sample size are being allocated 2:1 to receive either the bioengineered cornea (n=10), or an AM transplant (n=5), selected as the control treatment for trophic corneal ulcers in advanced stages (figure 1). In all trial subjects (n=20), study endpoints will be measured along a follow-up period of 24 months structured in 31 time points, that include three pre-implantation assessments, and 27 post-implantation evaluation visits (figure 2).

Trial design and stopping rules. Within phase I of the trial, the first five eligible patients were recruited sequentially with a safety period of 45 days between each other, receiving the bioengineered corneal graft (no randomisation). When all five subjects completed a 3 month post-implantation follow-up period, safety and feasibility data generated were analysed by the trial’s Data Safety Monitoring Committee (interim analysis), according to the trial’s stopping rule. Subject enrolment was re-activated within the phase II of the study, and the remaining fifteen patients that complete the study sample size are currently being recruited and randomly allocated 2:1 to receive either the bioengineered cornea (n=10), or aminotic membrane transplantation (n=5), selected as the control treatment for trophic corneal ulcers in advanced stages. All subjects recruited in the trial will complete a 24 months follow-up period.

{kind=link}
{kind=link}
Study visits and procedures. Clinical trial visits are structured in 31 time points, that include three pre-implantation visits (screening, randomization and pre-implantation) and 27 post-implantation evaluation visits according to a decreasing frequency rate: daily (week 1), weekly (month 1), fortnightly (months 2 and 3), monthly (months 4 to 12), and quarterly (months 13 to 24). The procedures and assessments performed are detailed for each visit.
The trial started in February 2014 and it is currently ongoing in ten Spanish hospitals, with the collaboration of the University of Granada and Cartuja Vision Clinic. The sponsor of the trial is the Andalusian Initiative for Advanced Therapies (IATA), with the support of the Andalusian Public Foundation Progress and Health , acting as the coordinating and advisory entity, and is responsible for handling clinical trial administrative authorizations and regulatory affairs, providing study supplies, independent data and safety monitoring, centralised manufacturing supervision, as well as general coordination and daily operational management of the trial for all of the participating sites.
Trial status
At the time of submitting this manuscript, phase I of the clinical trial had finished and the safety data obtained from the first five patients within the first 3 months of follow-up (all receiving the bioengineered corneal graft) had been evaluated by the Trial Data Safety Monitoring Committee (DSMC). After the trial’s DSMC approval in April 2016, phase II patient recruitment was re-activated, with seven patients implanted so far (table 1). The first patient of the clinical trial was included in January 2014, and enrollment period will be open until sample size (n=20) is completed, in June 2018 or earlier. Therefore, the trial is expected to finalise in June 2020, when the last patient included concludes its 24 months’ follow-up.
Clinical trial status
Outcomes
The main objective of the trial is to evaluate the safety, feasibility as well as evidence of clinical efficacy, of a model of human allogeneic nanostructured lamellar anterior cornea, in patients with severe trophic corneal ulcers for which there is currently no effective therapeutic alternative. Study variables regarding safety, feasibility and efficacy are measured as appropriate during the follow-up visits (figure 2). Safety and feasibility end-points are: adverse events (AEs) and serious adverse events (SAEs) causally related with study interventions or procedures (including marked changes of vital signs, ocular and physical examinations and laboratory test results); local, regional or systemic infections related with the implant; induced chronic ocular complications graded as described by Sotozono et al.28 As efficacy end-points: integrity, detachment or reabsorption of the bioengineered cornea/AM implant; ulcer persistency or relapse and corneal stromal repair; visual acuity; corneal transparency; quality of life (EQ-5); tear function (TBUT and Schirmer); SICCA ocular staining score29 and touch corneal sensitivity (Cochet-Bonnet esthesiometer). Additionally, the trial includes an in vivo confocal microscopy (IVCM) analysis of the grafted bioengineered cornea (and AM) using a Heidelberg Retina Tomograph equipped with a Rostock Corneal Module (HRT-RCM) for the micro-characterisation of the epithelium, the stroma and stromal nerves, as well as the corneoscleral limbus.
Random allocation
As sample size is small, block randomisation is applied to reduce bias and achieve balance in the allocation of participants to treatment arms. Randomisation list was computer generated by the Sponsor, and allocation is kept concealed for the research teams recruiting patients. In patients with injuries that meet criteria for treatment in both eyes, for ethical reasons the implant is applied only in one of them, the one with the worst prognosis according to the ophthalmologist’s criterion. The other eye may be treated with standard treatment, outside the context of the clinical trial.
Trial interventions
In both allocation groups, the surgical procedure is performed using topical or local (peribulbar or subtenoniane) anaesthesia. All patients allocated to the experimental arm are implanted with a bioengineered human allogeneic anterior lamellar cornea, obtained as described previously. The tissue-engineered implant covers the corneal scarring or defect as a graft, after debridement and preparation of the injury bed performing a 50–100 µm-thick keratectomy. The bioengineered corneal graft is implanted in the corneal lesion with the epithelial surface facing up, using interrupted 10–0 nylon sutures. Corneal sutures are removed 3–5 weeks after the implantation. Patients allocated to the control arm receive AM transplantation using a mixed graft/patch technique, named the sandwich technique, which is applied to cover the area of the corneal lesion after debridement and preparation of the injury bed.30
During the post-implantation follow-up, patients receive routine medical care based on existing clinical protocols for AM and corneal transplants procedures, as indicated in table 2.
Concomitant medication
Manufacturing of bioengineered human allogeneic anterior corneas
Bioengineered corneas are manufactured at the GMP facility of the Cell Production & Tissue Engineering Unit at University Hospital Complex of Granada (Virgen de las Nieves Hospital). Cultured cells are obtained from ex vivo cultures of corneoscleral limbus biopsies from cadaveric human donors. As there is no consensus on the indication of HLA-matching in corneal transplantation,31 and topic immunosuppressant are used after implantation, no HLA-matching criteria were considered in the selection of the sample. Tissue from donor corneoscleral rings is screened for transmittable diseases, washed and mechanically divided into limbus and central cornea. Limbal epithelial cells and stromal fibroblasts are then isolated and cultured in a suitable media until they can be cryopreserved and quality controls be performed. Bioengineered cornea is constructed by sequential culture techniques using a biodegradable scaffold based on a mixture of 0.1% agarose and fibrin (provided from human plasma that is coagulated in combination with the agarose and other elements). Corneal fibroblasts are first kept in culture within the biomaterial. Limbal epithelial cells are then seeded on top, and maintained in culture (the last culture sequence in an air-liquid interface). Once generated, corneal equivalents are subjected to plastic compression for partial dehydration (approximately 80%), or nanostructuration (patent P200930625 and P200930943). Strict quality control tests are performed including: viability, sterility, microbiological staining, mycoplasma, karyotype, fingerprint, virus culture, endotoxin and Chlamydia.14 32 Within the following 6 hours after manufacturing the product is transported at a controlled temperature (0°C to 8°C) to the hospital where it is programmed to be implanted.
Procurement and preparation of amniotic membrane
The innermost layer of donor placenta, the AM, is procured in sterile conditions, packed and cryopreserved (−75 to −85°C) under standardised operating guidelines by the Andalusian Public Health System Biobank.
Selection and enrollment
Criteria for patient eligibility are detailed in box. Subjects are selected among ≥18 years old patients presenting a trophic corneal ulcer grade three in Mackie’s classification,33 refractory to conventional treatment, or with sequelae of previous ulcers (such as stromal fibrosis). A target sample size of 20 subjects, entering the emergency units, or who are regularly followed-up as outpatients in the Ophthalmology Units of the participating sites, are being recruited.
Inclusion and exclusion criteria
Inclusion criteria
Participants meeting all the following criteria will be included:
Man or woman aged≥18, with no upper age limit.
Patients that give their informed consent for study participation.
Stage 3 Mackie corneal ulcers that do not respond to conventional medical treatment, or patients having undergone previous stage 3 Mackie corneal ulcers,33 currently suffering sequelae such as stromal fibrosis or corneal thinning, having no effective therapeutic alternative.
Stromal involvement, not reaching the Descemet membrane. Central or peripheral localization.
Minimum duration of the disease causing the corneal ulcer: 6 weeks.
No active ocular infection.
Patients with normal laboratory parameters as defined by: Leukocytes≥3000 cells/µL; Neutrophils≥1500 cells/µL; Platelets≥100 billion/L; AST/ALT≤1.5 ULN; Creatinine≤1.5 mg/dL.
Exclusion criteria
Participants meeting one or more of the following criteria will be excluded:
Absence of stromal involvement.
Good response to standard medical treatments for corneal disease in less than 3 to 5 weeks.
Bullous keratopathy or other endothelial decompensations.
Active ocular infection.
Positive serology to HBV, HCV, HIV or any other pathology that may interfere with correct patient follow-up.
Pregnant or breast-feeding women or childbearing-age women that do not consent the use of contraceptive methods approved in the protocol.
Medical history of active neoplasia within the past 5 years.
Participation in other clinical trials in 3 months previous to inclusion, or in the previous 5 years for trials with advanced therapies.
All candidates meeting selection criteria go through a standardised selection process, receiving all information about the study and the investigational product. The purpose, procedures and potential risks and benefits of the study are also explained thoroughly to the participants by study practitioners. All patients agreeing to participate in the clinical trial must provide written informed consent before undergoing any study-related procedures. A written approval consent form was also obtained from the legal representatives of the donors according to the applicable regulations. Patients are engaged to complete study follow-up through an optimised medical attention. Trial subjects have the right to withdraw consent for study participation at any time, and for any reason, or for no reason at all. Furthermore, consent removal is the only withdrawal criteria accepted by the protocol, once the patient has received the implant. In any other case, even if for any reason procedures programmed by the protocol cannot be fulfilled, the patient will complete follow-up visits to meet safety objectives. Withdrawn patients will not be substituted.
Data Safety Monitoring Committee
An independent DSMC was created as an advisory board to guarantee correct patient recruitment and withdrawal, to advice on any safety issues, to evaluate safety and feasibility data of the trial interim analysis (phase I, safety data throughout 3 months of follow-up), and to be involved in any decisions ensuring the compliance of Good Clinical Practice (or GCP), the protocol procedures and applicable regulations. The DSMC consists of four members, independent from sponsor or clinical teams, including two expert consultants in the field of corneal disease, a clinical methodologist and a scientific adviser.
Follow-up protocol
After receiving the implant, all patients enrolled in both phases of the trial are followed-up for 24 months according to planned visits (figure 2) by clinical ophthalmologist at the participating sites, ensuring that essential clinical, analytical and exploratory data about safety and efficacy are registered in the study’s Case Report Form (CRF). Standard methods for the evaluation of the anterior pole of the eye are used (clinical evaluation, eye staining tests, slit lamp, OCT, Cochet-Bonnet esthesiometer, etc.), as well as periodic exams with IVCM, in hospitals equipped with an HRT-RCM.
Clinical trial visits are structured in 31 time points, that include three pre-implantation visits (screening, randomization and pre-implantation) and 27 post-implantation follow-up assessment visits according to a decreasing frequency rate: patients receiving the bioengineered corneal graft have daily visits during week 1 (hospitalisation is required for at least 1 day). Patients receiving the AM transplantation have the first visit 24 hours after the implant, then every 48 hours during week 1 (hospitalisation not required). Then, follow-up scheduled visits include weekly visits during month 1, fortnightly visits in months 2 and 3, monthly visits in months 4 to 12, and quarterly visits during the second year of follow-up. The procedures and assessments performed in each visit are listed in figure 2.
Safety and adverse events assessments
In accordance with GCP, all AEs and SAEs occurring during the study, observed by the investigator or reported by the patients, whether or not attributed to the investigational medicinal product, are carefully monitored and recorded on the trial CRF. Also, investigators must report to the sponsor any SAEs within 24 hours of the onset. Adverse events related to the study interventions or procedures are followed-up until satisfactory resolution or stabilisation.
Safety assessments include the evaluation of the safety of the procedure (including AEs occurred within 24 hours after the intervention), and the evaluation of the safety of the product (including AEs occurred during the 24 months follow-up period, as well as adverse reactions occurred subsequently). AEs are classified on the basis of MedDRA terminology and summarised for each treatment arm. AEs incidence rates will be summarised by System Organ Class (SOC), Preferred Term (PT), severity and relationship to the intervention. Safety analysis will include the comparison of the percentage of patients presenting AEs (and SAEs) related or not with the interventions and the most frequent AEs in each treatment arm.
The causality of AEs with the study intervention is assessed by the principal investigator, and re-evaluated by a qualified person responsible for pharmacovigilance appointed by the trial’s sponsor. Furthermore, the study’s DSMC is responsible for reviewing the accumulated safety and efficacy data, when appropriate, and for making recommendations to the sponsor concerning the continuation, amendment and termination of the trial.
Additionally, the trial will be stopped if any of the following circumstances were met: severe toxicity or infections related with the bioengineered corneal implant in three or more patients; or mortality related with the artificial corneal graft in one or more patients.
Sample size calculation
Trial sample size has not been obtained from statistical calculations. It is based on epidemiological and clinical data, seeking to obtain significant initial information about safety and feasibility, minimising at the same time drug exposure, as this is the first trial in humans with a model of bioengineered allogeneic anterior cornea containing two different human cell types.
Data management and statistical analysis
CRF data will be manually entered in the study database, through double data entry and discrepancy management procedures. A query plan will be executed for missing and out of range values. Sponsor and investigators will have access to the final trial dataset. Safety and feasibility evaluation will be performed with intention-to-treat (ITT) analysis. Per-protocol analysis (PPA) will be used for efficacy assessments. The ITT analysis will include all patients who agreed to participate in the study, signed informed consent and were randomised. PPA will include patients who were randomised, received the assigned intervention, excluding those who registered severe protocol deviations (violations). Statistical analysis will take into account the center-effect issue by including the site as an exploratory variable in the model. If some study centres have very few subjects, a pooling strategy will be employed to combine the small centres together.
Data monitoring
With the final purpose of protecting patients and guaranteeing data quality and trial integrity, a clinical monitor or Clinical Research Associate (CRA), appointed by the trial sponsor, takes responsibility for supervising the study progress in each investigational site, involving periodical on-site visits as well as centralised supervision activities throughout the duration of the study. The aim of monitoring is to ensure that investigators are following the protocol, complying with regulatory and GCP standards, and collecting and reporting quality data.
Ethics and dissemination
The trial protocol obtained the authorisation of the Spanish Agency of Medicines and Medical Devices and the Coordinating Institutional Review Board of Biomedical Research in Andalusia (Referral Ethics Committee), which gathered the approval from local ethics committees in all the participating hospitals. All substantial amendments to the original approved documents also obtained further approval from the corresponding Institutional Review Board (IRB) and Regulatory Authority. The clinical trial protocol has been subject to seven amendments so far, including minor changes in patient eligibility criteria, follow-up procedures, as well as in the protocol’s randomization ratio. The study presented describes the current approved protocol at the time of submission: Amendment 7, 27th December 2016. Moreover, sponsor and investigators ensure that this trial is conducted in accordance with the principles of the Declaration of Helsinki, International Conference Harmonisation Guidelines (ICH) for GCP and in full conformity with applicable regulations. The sponsor, investigators and other staff involved in the trial ensure that participant’s confidentiality is preserved.
All items from the WHO Trial Registration Data Set were registered in the publicly accessible databases ClinicalTrials.gov (NCT01765244, January 2013) and the EU Clinical Trial Register (2010-024290-40, December 2012). On completion of the trial, manuscripts with the results of phases I and II of the study will be published in a peer-reviewed journal and presented in national and international conferences. Authors will be selected based on their contribution to the results.
The SPIRIT statement34 has been observed in the publication of this study protocol and the CONSORT guidelines35 will be guaranteed when publishing the study results in clinical journals and conferences.
Discussion
There is a clear need to develop a corneal substitute that can optimally repair the human cornea when it is damaged. In this context, fibrin-agarose corneal substitutes combining allogeneic stem cells and biomaterials might mimic the human native anterior cornea in terms of optical, mechanical and biological behaviour. To evaluate the safety in humans of this tissue engineered advanced therapy, we designed a phase I – II clinical trial recruiting patients suffering from severe trophic corneal ulcers. The intervention is based on the implantation of a bioengineered human allogeneic nanostructured lamellar anterior cornea after performing an anterior lamellar keratectomy to remove the damaged corneal tissue. Severe trophic corneal ulcers usually require aggressive treatments such as the use of glues, corneal transplants or AM grafts to preserve the anatomical integrity of the cornea.26 27 Thus, fibrin-agarose scaffolds could also be applied here to preserve the corneal structure, adding growth factors from fibrin, allowing us to evaluate the safety of this new advanced therapy for the first time in human patients.
We have designed the trial with wide eligibility criteria for an easier screening and recruitment, making the study more feasible and economical, especially in the context of an investigator-driven, publicly funded clinical trial. Although this strategy may provide a heterogeneous sample, we believe it will not interfere with the comparison of the main safety outcome, as we are measuring important prognostic variables at baseline and throughout follow-up to allow for a precise discrimination of causality of the events. The special characteristics of the trial interventions impeded the possibility of blinding, which is a limitation to the study design. To avoid evaluation bias both allocation groups follow-up procedures schedules are equivalent, standardised grading systems are used when possible for objective outcome measurements and statistical analysis will be performed by blinded data analysts. Moreover, this study has been designed as a small size randomised controlled trial due to limitations in the current expiration period of the investigational product, which restricted site selection process. The reason to use AM as control is that it is commonly applied to treat severe corneal ulcers. Moreover, the implantation technique of AM is based on sutures, similar to the one employed for the fibrin-agarose scaffold implantation, in comparison to the use of glues, such as fibrin-based glue or cyanoacrylate. To our knowledge, this is the first report showing an investigator-driven multicentre randomised clinical trial evaluating bioengineered corneal implants combining two types of allogeneic cells (limbal epithelial cells and corneal fibroblasts). The design, management and coordination efforts invested on this trial may be of special interest to serve as a model for emerging multicentre non-commercial clinical trials on tissue engineered products.
Once the safety of this advanced therapy is established in humans, the final goal will be to evaluate its efficacy to restore the whole anterior cornea in patients with LSCD that associates a damage in the corneal stroma (ie, corneal scarring). This will be carried out in a future phase III clinical trial, if the current one demonstrates the safety and feasibility of the evaluated treatment. Thus, the results of the present trial are expected to envision a new therapeutic alternative for the treatment of severe corneal diseases that require regeneration of the limbal epithelial stem cell population and the corneal stroma, using tissue engineering advanced therapies.
Acknowledgments
To all involved in the trial, the sponsor, the clinical teams in the participating sites, the manufacturer, the Biobank of the Andalusian Public Health System, the investigators, and particularly the patients.
References
Footnotes
Participating institutions Andalusian Initiative for Advanced Therapies, Seville (Sponsor, coordination and advisory entity); University of Granada (Scientific coordination); Biobank of the Andalusian Public Health System (Supply of cadaveric human scleral-corneal limbus); Cell Production & Tissue Engineering in University Hospital Complex of Granada, Virgen de las Nieves Hospital (Manufacturer of investigational medicinal product); Clinical sites (Patient recruitment, follow-up and data collection): University Hospital Complex of Granada, San Cecilio and Virgen de las Nieves Hospitals (Sites 1), Virgen Macarena University Hospital-Seville (Site 2), Virgen del Rocio University Hospital-Seville (Site 3), Puerta del Mar University Hospital-Cadiz (Site 4), Reina Sofia University Hospital-Córdoba (Site 5), La Arruzafa Hospital-Cordoba (Site 6), Costa del Sol Hospital-Marbella (Site 7), Nª Sª de Valme Hospital-Seville (Site 8), University Regional Hospital Malaga (Site 9), San Juan de Dios Hospital-Seville (Site 10); Cartuja Vision Clinic (In vivo confocal microscopy evaluations).
Contributors MGA, MCGG, MA, RM, NC, SM, ARG, LPF, ALM, IG, AC, GC: were involved in conception, investigational product development and trial design. MGA, MCGG, MA, SAS, JMA: were involved in drafting of the manuscript. All authors contributed to critical revision of the manuscript and read and approved the final version.
Funding This Clinical trial is investigator-driven and partially supported by a research grant for the promotion of investigator-driven clinical research from the Spanish Ministry of Health, Social Policy and Equity (Grant Number: EC10-285). This study is also supported by the National Plan for Scientific Research, Development and Innovation from the Spanish Ministry of Economy and Competitiveness (Institute of Health Carlos III), grant code: FIS PI14/0955 (co-financed by FEDER funds, European Union). Finally, the study is supported too by the Regional Ministry of Health of Andalusia, who finances the costs incurred by participating hospitals, and the Andalusian Initiative for Advanced Therapies. The Andalusian Initiative for Advanced Therapies, through the Andalusian Progress and Health Public Foundation, assumes the roles and responsibilities of sponsoring this clinical trial.
Competing interests Dr. González-Andrades, Dr. Alaminos and Dr. Campos are inventors of issued patents P200930625 and P200930943, broadly relevant to the work. Remaining authors declare that they have no competing interests.
Patient consent Obtained.
Ethics approval Comité Coordinador de Ética de la Investigación Biomédica de Andalucía.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data sharing is not applicable to this article as no datasets were yet generated or analysed.