Article Text

Abstract
Introduction While some evidence exists that real-time remote symptom monitoring devices can decrease morbidity and prevent unplanned admissions in oncology patients, overall, these studies have significant methodological weaknesses. The electronic Symptom Management using the Advanced Symptom Management System (ASyMS) Remote Technology (eSMART) study is designed to specifically address these weaknesses with an appropriately powered, repeated-measures, parallel-group stratified randomised controlled trial of oncology patients.
Methods and analysis A total of 1108 patients scheduled to commence first-line chemotherapy (CTX) for breast, colorectal or haematological cancer will be recruited from multiple sites across five European countries.
Patients will be randomised (1:1) to the ASyMS intervention (intervention group) or to standard care currently available at each site (control group). Patients in the control and intervention groups will complete a demographic and clinical questionnaire, as well as a set of valid and reliable electronic patient-reported outcome measures at enrolment, after each of their CTX cycles (up to a maximum of six cycles) and at 3, 6, 9 and 12 months after completion of their sixth cycle of CTX. Outcomes that will be assessed include symptom burden (primary outcome), quality of life, supportive care needs, anxiety, self-care self-efficacy, work limitations and cost effectiveness and, from a health professional perspective, changes in clinical practice (secondary outcomes).
Ethics and dissemination Ethical approval will be obtained prior to the implementation of all major study amendments. Applications will be submitted to all of the ethics committees that granted initial approval.
eSMART received approval from the relevant ethics committees at all of the clinical sites across the five participating countries. In collaboration with the European Cancer Patient Coalition (ECPC), the trial results will be disseminated through publications in scientific journals, presentations at international conferences, and postings on the eSMART website and other relevant clinician and consumer websites; establishment of an eSMART website (www.esmartproject.eu) with publicly accessible general information; creation of an eSMART Twitter Handle, and production of a toolkit for implementing/utilising the ASyMS technology in a variety of clinical practices and other transferable health care contexts.
Trial registration number NCT02356081.
- cancer
- chemotherapy
- information technology
- symptom management
- randomised controlled trial
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Suitably powered multicentre (multiple clinical sites across five European countries) randomised controlled trial of the efficacy of remote technology to manage symptoms in oncology patients.
Measurement of patient outcomes during active treatment through to and inclusive of a survivorship period (ie, 1 year post treatment).
Potential bias associated with the possibility that only technologically confident patients will agree to take part and the limited number of European countries involved in the study.
Introduction
Recent advances in chemotherapy (CTX) have resulted in considerable increases in overall cancer survival rates.1–4 However, this treatment is associated with significant5 6 and occasionally life-threatening side effects.7–9 In addition, CTX-related symptoms can lead to poor treatment adherence,10 increased hospitalisations,7–9 11 12 increased costs7–9 11 12 and impaired quality of life (QoL).13–15 CTX-related symptoms occur not only during active treatment but can persist into survivorship.6 16 17
While symptom assessment is an essential component of cancer care, regular and systematic approaches to symptom assessment are lacking.18 19 Currently, identification of CTX-related symptoms primarily relies on retrospective patient recall, which, apart from being prone to recall bias and inaccuracy,20 may be of limited benefit because delays in reports of clinically significant and potentially life-threatening symptoms impair effective symptom management.21 Ineffective symptom management results in disruptions to health-related QoL (HR-QoL) and increases supportive care needs.22 These unfavourable outcomes have negative effects on patients and their family caregivers.23 Effective symptom assessment and management is further compounded by the transition of cancer services from traditional inpatient care towards care delivered within local settings.24 This shift in care delivery means that many patients are required to engage in self-care activities to prevent or reduce the severity of CTX-related symptoms24 and make important treatment decisions without their clinicians.25 The use of innovative technological systems provides an affordable solution to the increasing demands placed on acute care through the delivery of care in the home and rural settings.26 27 Such remote monitoring systems facilitate real-time communication between patients and their clinicians26 and in recent years there has been a rapid increase in the number of these systems being used in the cancer setting,28 many of which require further development in order to enhance their usability and clinical integration.
One of the more advanced remote monitoring systems available to assess and manage CTX toxicities is the Advanced Symptom Management System (ASyMS), which was developed by researchers, cancer clinicians and people with cancer.29–36 ASyMS is a mobile phone-based remote monitoring system that enables the ‘real-time’ monitoring of patients’ symptoms through use of patient-reported outcome measures (PROMs). The use of PROMs is advocated as an effective way to identify aspects of a patient’s health status,37 enhance management of CTX-related symptoms, alleviate patient anxiety and promote self-care self-efficacy.38 Combined with technology-driven interventions that are able to capture symptom data in real time, electronic PROMs (ePROMs) allow for rapid clinical decision-making and interventions to improve patient outcomes.39 In addition, they enable the delivery of quality care irrespective of distance27 and economic or cultural contexts.
Complementary to such technological innovations is the advent of predictive risk models (PRMs) which enable care to be effectively triaged and clinicians to employ a preventative and anticipatory model of care through identification of patients at greatest risk for CTX-related symptoms.40 Collectively, the use of technologically driven, real-time symptom monitoring and predictive risk modelling will allow for timely, high-quality, person-centred supportive care.
Recent evidence demonstrates that remote symptom monitoring through patient report leads to clear clinical benefits.41 The clinical impact of these interventions includes decreased morbidity, improved HR-QoL and a reduction in hospital admissions in oncology patients.41 However, the concept is still relatively novel with very few studies conducted in cancer settings.42 Methodological weaknesses have been identified and none included a health economic analysis.42 The electronic Symptom Management using the Advanced Symptom Management System Remote Technology (eSMART) study is designed to address these issues with an appropriately powered, randomised controlled trial (RCT) which will demonstrate the effects of the ASyMS intervention on key patient outcomes and delivery of care provided to oncology patients during and after CTX.
Methods and analysis
eSMART is a two-part, pragmatic, 5-year RCT being conducted in multiple sites across five countries (ie, Austria, Greece, Ireland, Norway and the UK). Part 2, the protocol that is described in this paper, is the RCT. Part 1 was concerned with the necessary preparatory work required for the RCT (including scoping review, clinical algorithm refinement, patient/clinical advisory group discussions, translation and linguistic validation of all study materials and feasibility testing of the ASyMS technology). The eSMART study is sponsored and led by the University of Surrey in the UK and is registered on ClinicalTrials.gov (NCT02356081).
The primary aim of eSMART is to evaluate the short-term and long-term impact of the ASyMS technology on patient-reported outcomes in oncology patients receiving first-line CTX for breast cancer, colorectal cancer (CRC) and haematological (ie, Hodgkin’s disease (HD), non-Hodgkin's lymphoma (NHL)) cancer. In addition, eSMART will evaluate the cost-effectiveness of the ASyMS technology, as well as changes in clinical practice that may follow its full deployment in different European healthcare settings. The primary and secondary study objectives and their associated outcome measures (ePROMs) are as follows.
Primary objective
To determine whether, compared with standard care, the ASyMS intervention can lead to reduced symptom burden during active CTX for breast cancer, CRC, HD or NHL as evidenced by a statistically significantly lower total Memorial Symptom Assessment Scale (MSAS)43 score over six CTX cycles. The MSAS is a valid and reliable PROM43 that evaluates 32 physical and psychological symptoms according to their frequency, severity and distress/bother to the patient in the past week.
Secondary objectives
To determine whether, compared with standard care, the ASyMS intervention can lead to the following outcomes in patients receiving CTX for breast cancer, CRC, HD or NHL:
Reduced symptom burden during active CTX as evidenced by statistically significantly lower total MSAS scores at prespecified time points and at 1 year following the intervention.
Reduced symptom burden at mid-CTX cycle time point (ie, at a time point when symptom burden is known to reach its peak), as evidenced by a statistically significantly lower total MSAS score.
Increased HR-QoL (ie, statistically significantly higher scores on overall and HR-QoL domains of the Functional Assessment of Cancer Therapy-General44 during active CTX and/or at the 1 year follow-up.
Reduced supportive care needs (ie, statistically significantly lower scores on supportive care needs domains of the Supportive Care Needs Survey-Short Form 3445 during active CTX and/or at the 1 year follow-up.
Reduced anxiety (ie, statistically significantly lower scores on the state and/or trait anxiety domains of the State-Trait Anxiety Inventory-Revised46 during active CTX and/or at the 1 year follow-up.
Improved self-care self-efficacy (ie, statistically significantly higher self-efficacy scores on the Communication and Attitudinal Self-Efficacy scale for cancer47 during active CTX and/or at the 1 year follow-up.
Fewer work limitations (ie, statistically significantly lower work limitations scores on the Work Limitations Questionnaire 48 during active CTX and/or at the 1 year follow-up.
In addition, the following outcomes will be evaluated:
The cost-effectiveness of the ASyMS intervention for the management of CTX-related symptoms by combining resource use data with quality-adjusted life-years measured with the EuroQol 5-Dimensions49 and use of the Client Services Receipt Inventory.50
Changes in clinical practice as a result of the ASyMS intervention by promoting an anticipatory and preventative model of care that enables more care to be delivered in local settings.
PRMs to predict CTX-related symptoms in patients with breast cancer, CRC, HD or NHL by combining demographic, clinical, social and health service data collected from previous studies, as well as the current study, to inform the predictions made. This component will move beyond traditional approaches to manage symptoms in oncology patients receiving CTX to more tailored and anticipatory approaches.
Setting
Patients (n=1108) will be recruited from multiple sites across five European countries (see online supplementary appendix 1 for details).
Patient screening and recruitment
At each site, eligible patients (see Box 1 for eligibility criteria) will be identified by the local site Principal Investigator or other members of the local clinical team. Patients will be recruited from outpatient and/or inpatient oncology settings prior to the initiation of CTX.
Eligibility criteria
Patients will be included in the RCT if they are
Adults (≥18 years)
Diagnosed with breast cancer, CRC, HD or NHL
Scheduled to receive first-line CTX or (if previous CTX has been received) scheduled to receive CTX for the first time in the last five years
Scheduled to receive 2-weekly, 3-weekly or 4-weekly CTX protocols (ie, CTX administered every 14, 21 or 28 days, respectively)
Scheduled to receive a minimum of three cycles of CTX
Physically/psychologically fit to participate in the study
Able to understand and communicate in the respective language
Patients will be excluded from the RCT if they are
Diagnosed with a distant metastasis in the case of breast cancer or CRC
Experiencing B symptoms in the context of an HD or NHL diagnosis
Scheduled to receive concurrent radiotherapy
Scheduled to receive weekly CTX
Diagnosed with recurrent cancer or another type of cancer within 5 years prior to recruitment (with the exception of non-melanoma skin cancer)
Patients who have CTX within the previous five years for any medical reason
Unable to provide written informed consent
CRC, colorectal cancer; CTX, chemotherapy; HD, Hodgkin’s disease; NHL, non-Hodgkin's lymphoma; RCT, randomised controlled trial.
Patient recruitment
Clinical staff at each site and/or dedicated research staff will assist with the recruitment of patients. Patients will be given a sufficient period of time to consider participation and they will be advised that they can discuss the study with any significant others and/or health professionals prior to making a final decision. If patients agree to participate, they will be asked to attend the clinic prior to their prearranged appointment for CTX (enrolment visit). During the enrolment visit, written informed consent will be obtained (see online supplementary appendix 2).
Randomisation
Following the consent procedure, randomisation will be performed remotely and independently by the Surrey Clinical Trials Unit (CTU) using the Promasys system (OmniComm Systems). Research staff at each site will log into the randomisation site. Eligible patients will be randomised via a web-based electronic case report form and will be stratified by site and type of cancer. These steps will ensure that bias in treatment assignment, specifically selection bias, allocation concealment bias and confounding bias, will be eliminated. This allocation will determine whether the patient is allocated to the mid-CTX measurement (30% of total study sample). Information concerning patient eligibility, patient unique ID number, date of randomisation, allocation group and written informed consent will be recorded in the Promasys system. Due to the nature of the intervention, blinding is not possible. However, to mitigate the adverse effects of blinding bias, patient information sheets (see online supplementary appendix 3) will be deliberately produced to avoid any reference to ‘intervention group’ or ‘control group’ as this information could discourage patients allocated to the control group from taking part. Instead, all patients will be informed that they will be randomly allocated to one of two different methods of symptom management during CTX (ie, either ‘mobile phone group’ or ‘normal care group’). In addition, patients will be blinded to study hypotheses.
Prior to advising patients about the outcome of the randomisation process, all patients will be asked to complete a set of ePROMs on a tablet personal computer (PC) or desktop PC. If patients are allocated to the intervention group, the research staff will provide a brief overview of study procedures and train the patient to use the ASyMS technology prior to commencement of CTX. If patients are allocated to the control group, the research staff will provide a brief overview of study procedures.
Intervention
The ASyMS intervention uses mobile phone technology to enable real-time monitoring of patients’ CTX-related symptoms (figure 1A). The core component of the ASyMS intervention is the mobile phone device (ie, ASyMS patient handset shown in figure 1B), which contains an electronic version of the ASyMS symptom questionnaire (CTX Toxicity Self-Assessment Questionnaire (CTAQ)). The CTAQ assesses 10 CTX-related symptoms (ie, feeling sick, being sick, diarrhoea, constipation, sore mouth and/or throat, paraesthesias, sore hands and/or feet, flu-like symptoms/infection, tiredness, pain). An additional item is included to give the patient the option to report up to six further symptoms.
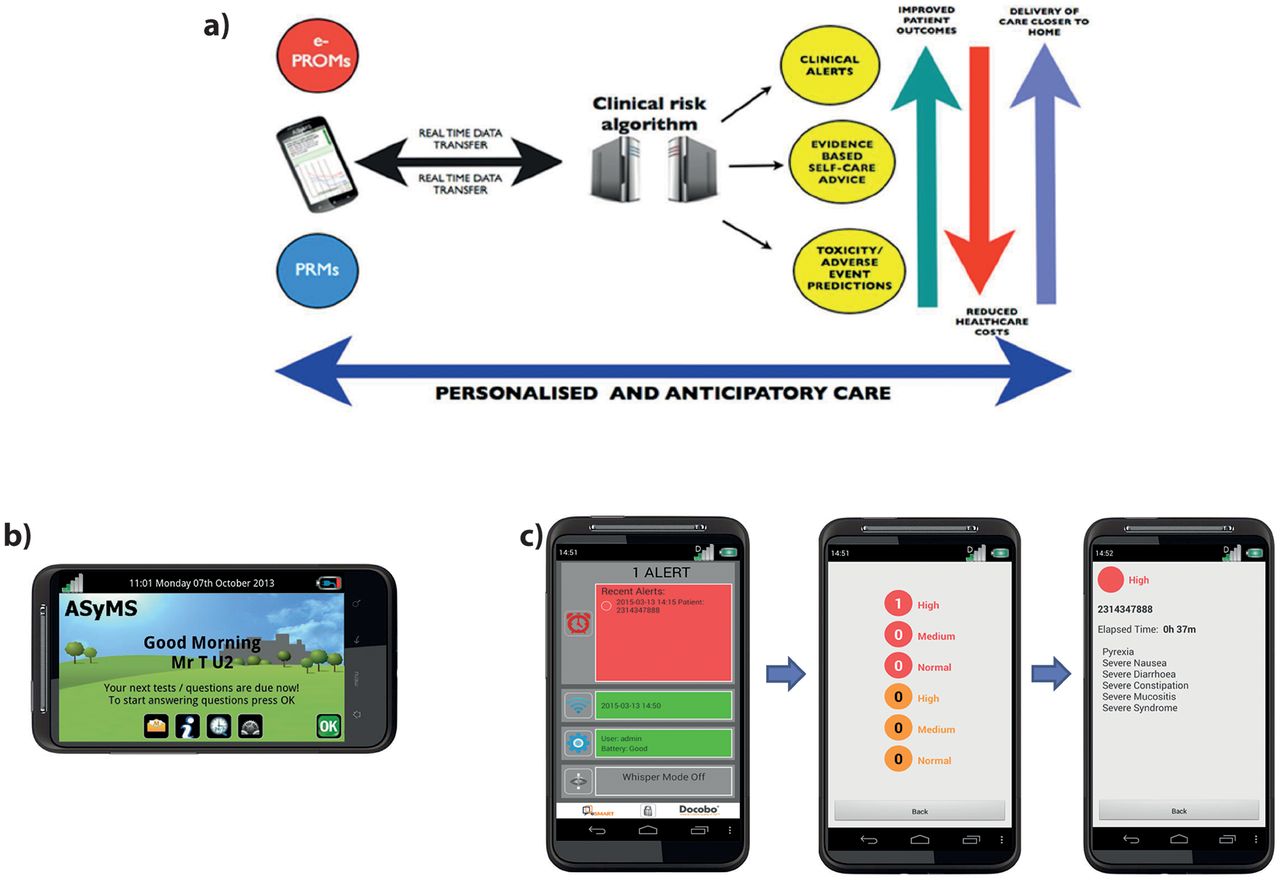
{kind=link}
Electronic Symptom Management using the Advanced Symptom Management System Remote Technology (eSMART) intervention: (A) Diagram illustrating the eSMART model of care. (B) Patient ASyMS device. Example of the home screen where the patient can access self-care advice, their daily symptom questionnaire and any text messages sent by clinician. (C) Clinician alert-handling process. These screenshots provide an example of the type of information available on the clinician handset, that is, severity of alert, patient ID and list of reported symptoms.
This ePROM evaluates three dimensions of CTX-related toxicity on separate response scales, namely incidence, severity and bother. Symptom occurrence is reported on a dichotomous scale (ie, yes/no). When the answer is ‘yes’, the patient is asked to rate the severity and bother of each symptom. A three-point scale (mild, moderate, severe) is used to evaluate symptom severity. The severity indicators have associated descriptors based on the Common Terminology Criteria for Adverse Events V.4.0.51 Mild symptoms are defined using CTC 1 criteria, moderate symptoms CTC 2 and severe symptoms CTC 3. This approach ensures that ratings of symptom severity are easily translated into routine practice and incorporated into current management guidelines. A four-point scale (not at all, a little, quite a bit, very much) evaluates how much a patient is bothered by the symptom. All symptoms will be rated using a recall period of the past 24 hours. The CTAQ symptom questionnaire was used in previous ASyMS studies.33 The validity and reliability of this tool was demonstrated in oncology patients receiving CTX.33
Prior to completing the ePROM, patients will be asked to indicate whether they are at home or in hospital, they will complete the CTAQ symptom questionnaire on the ASyMS patient handset and take their temperature with a tympanic electronic thermometer. Patients will enter this value into the handset once daily and at any time they feel unwell after each of their CTX cycles (up to a maximum of six cycles). Patients involved in previous ASyMS studies did not find this process burdensome and adherence across patient populations was high.36 52Patients will immediately receive automated, evidence-based self-care advice based on their symptom reports. In addition, the ASyMS patient handsets will provide access to a self-care library, symptom graphs (detailing trends in individual symptoms experienced) and contact numbers for care teams and patient support organisations in their country.
The patients’ ‘real-time’ symptom information will be sent automatically via a secured connection to Docobo’s (software provider) secure server. Patients will be advised via the ASyMS patient handset if their symptom data were sent successfully to the server. If the patient is at home and the incoming symptom reports are of clinical concern (eg, a developing infection), the server software will generate alerts that will be sent to the patients’ clinician. Two levels of alerts will be generated if symptoms require intervention. The first of these, an amber alert (to be addressed within 8 hours), will be for symptoms that are bordering on becoming problematic and would be responsive to early interventions. In such cases, clinicians will make a clinical judgement as to whether or not to call the patient following a review of the patient’s symptom information on the ASyMS website (ie, the patient’s ‘real-time’ symptom reports, patient symptom graphs and the 28-day-view patient display). A red alert (to be addressed within 30 min) will be triggered for symptoms that are severe or life-threatening (eg, fever). These time frames, within which an alert is to be answered, are designed to facilitate timely clinical intervention while also being manageable for the clinicians.
Clinicians will receive alerts on a dedicated ASyMS clinician handset (ie, a specialised mobile phone that the clinician responsible for handling alerts on a given shift (‘alert handler’) will carry with them at all times). All clinical sites will be provided with centrally generated, site-specific usernames and passwords that will allow clinicians access to the ASyMS web-based system. When an alert is triggered, the ASyMS clinician handset will play an audio attention prompt in the form of a repetitive, high-pitched ringing. Alert handlers will be able to identify which patient triggered the alert by tapping the relevant alert icon on the ASyMS clinician handset to reveal the type of incoming alert (amber or red), the patient study ID number, the time elapsed since the alert was received and, when available, a list of symptoms that triggered the specific alert (figure 1C). This information will allow the alert handler to match the alert to the patient on the secure eSMART website, where all patients’ symptom reports, demographic and clinical information, contact telephone numbers and addresses are viewable. On receipt of an alert, the clinician will view the patient’s ‘real-time’ symptom reports on a secure web page, before contacting the patient to initiate the appropriate intervention. Information on this secure website will include demographic and clinical information to allow clinicians to verify the patient’s identity and to assist in decision-making and subsequent interventions. Clinicians can use information stored on the eSMART website to conduct a clinical assessment with the patient. The alert handler will provide appropriate, standardised interventions using clinical algorithms which are based on international, national and local guidelines, and feedback from clinicians and patients. The alert handler will then document the actions/interventions performed, and sign off the alert on the eSMART website.
In addition to patient information being stored on the website for access during alert handling, a hard copy of the patient’s study ID number, name and contact details will be kept in a locked file at each site in case the eSMART server malfunctions or is unavailable. Thus, alert handlers will be able to contact the patient to assess their symptoms and intervene.
Training/education
Patient training (intervention group)
Patients allocated to the intervention group will be provided with the ASyMS patient handset and a tympanic thermometer. The research staff will instruct the patients on how to use the handset. If a patient experiences any problems with this procedure, additional training will be done until the patient feels comfortable using the ASyMS patient handset and they can successfully transmit their symptom information to the server. A manual for the handset containing instructions and contact numbers will be given to each patient. In the interest of patient safety, patients who will use the ASyMS intervention will be reminded that in cases where the technology fails, standard care will apply. For example, if symptoms are reported that will trigger an alert, before closing the questionnaire the patient will receive a message to inform them that their symptoms will be reviewed by a clinician and that they may receive a call within the time frame given. If patients do not receive any contact from the hospital (ie, in the case of technology failure) and feel that their symptoms are of concern, they are always advised at the outset of the study to use the standard care procedures.
Patients will be advised that they can still use their ASyMS handset if they are hospitalised. In this instance, no alerts will be triggered but patient data will be transferred to the server if internet connectivity is available. Patients in the intervention group will be informed about data collection procedures.
Patient training (control group)
Patients allocated to the control group will be informed about data collection procedures (see ‘Data collection and management’ section).
Clinician training on the intervention
Dedicated training sessions with members of each clinical team (local ‘alert handlers’) will take place prior to commencement of patient recruitment. In the interest of patient safety, clinicians will be reminded that in cases where the technology fails standard care will apply for patients in the intervention group throughout their participation in the study.
Data collection and management
Research design: Repeated measures, parallel group, stratified RCT.
Demographic and clinical data will be collected at the outset of the study. The outcome measures used in this study were selected following a review of the literature as advocated by the Medical Research Council framework for complex interventions.53–55 These PROMs were selected as the best available and most appropriate measures to address the primary and secondary objectives listed above.
Active treatment period
Irrespective of study condition, all patients will be asked to complete a set of ePROMs (primary and secondary) at enrolment (ie, prior to first CTX) and around the start of each subsequent CTX (between 2 days before and 1 day after CTX administration) up to a maximum of six cycles of CTX. In addition, a subsample (30%) of randomly selected patients from the intervention and control groups will be asked to complete a mid-cycle measurement (mid-CTX) of the primary outcome measure (ie, MSAS) at each cycle in order to capture additional data on symptom burden at this time. Depending on the duration of a given CTX cycle and patient availability, patients will be asked to complete the mid-CTX assessment between days 6 and 8 for 2-weekly protocols, between days 9 and 11 for 3-weekly protocols and between days 13 and 15 for 4-weekly protocols.
PROM data will be collected in the hospital, where patients will be asked to use a tablet PC or desktop PC each time they are required to complete the questionnaires. When required, research staff will assist patients to enter information on the tablet PC/PC. The tablet PC/PC will hold electronic versions of the PROMs questionnaires, allowing data to be transferred to the secure server. During active CTX, the data collection will coincide with patients’ visits for their next CTX cycle. To offer patients more flexibility, they may also have the option of completing PROM data via a secure web link sent to the patient’s email. Data collection of PROMs will take approximately 40–60 min; patients will be encouraged to take a break at any stage and will be regularly informed of their progress through the questionnaires. Should patients decide to take a break, the tablet PC/PC will automatically resume where they discontinued.
The data collection from the subset of patients who complete the mid-cycle MSAS measurements will be conducted by using either a secure web link for those patients who use email or through a telephone interview with the research staff. During this mid-cycle measurement, the research staff will record patient responses to the MSAS items on the same tablet PC/PC that will be used for patient self-reported data collection.
Follow-up period
On completion of a maximum of six cycles of CTX, both the intervention and control groups will be followed for 1 year to evaluate sustainability of the intervention effects. Follow-up data will be collected at prespecified time points every three months. A post-CTX ‘baseline assessment’ will take place after the end of the final CTX cycle. Subsequent assessments will take place at 3, 6, 9 and 12 months following this ‘baseline assessment’.
In order to reliably assess the sustained effects of the intervention, it is important that strategies are put in place to minimise the risk of attrition during the follow-up period. Therefore, a sequential mixed-mode design for data collection will be used to allow for flexibility in data collection and to control for non-respondent follow-up. PROM data will be collected in the hospital, where patients will be asked to use a tablet PC or PC each time they are required to complete the ePROMs. Patients will be given two alternative options if data cannot be collected in clinics using the tablet PC/PC: (1) internet surveys (sent via personalised emails) with automated reminders sent at 14 and 21 days post-due date to non-responders or (2) telephone-conducted interviews completed by research staff, who will enter data on a tablet PC/PC.
The use of sequential mixed-mode design for non-respondent follow-up may enhance perceptions of the importance of the research, increase response rates, be cost effective and reduce non-response bias with minimal effect on data quality.56
Sample size calculation
Sample size estimation was based on existing evidence of differences in total MSAS scores between intervention group and control group (ie, a difference between intervention and control groups of 1.45 – 1.30=0.15).57 Drawing on these data, a sample size estimation analysis indicated that for a difference in total MSAS score of 0.15 (SD=0.6) given an effect size of 0.25, with four repeated measures after enrolment and one enrolment measure, a sample of 776 patients (110 patients with haematological cancers and 333 patients with breast cancer and CRC) will provide 90% power for a two-sided 5% significance level.58 Allowing for an attrition rate of 30%, a total of 1108 patients will need to be recruited. During the postintervention follow-up period, it is assumed that an additional 30% of patients will dropout, giving a sample of 544 expected to complete the study 12 months post treatment.
In addition, to allow for a mid-CTX comparison of MSAS scores between intervention and control groups, a random 30% of the total sample recruited for the RCT (ie, n=334 (n=122 patients with CRC (61 intervention/61 control), n=122 patients with breast cancer (61 intervention/61 control) and n=90 patients with haematological cancer (45 intervention/45 control)) will be selected to provide data at this time point.
Data analysis plan
The statistical analysis of the data will be the responsibility of the Surrey CTU in conjunction with University of Dundee (please see the complete data analysis plan in online supplementary appendix 4). Demographic and clinical characteristics recorded at enrolment will be tabulated by treatment groups. Descriptive statistics will include n, mean, SD, median, minimum and maximum. All analyses will follow the guidance contained in the ICH E9 ‘Statistical Principles for Clinical Trials’ (http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002928.pdf). Analysis of these RCT data will be based on the intention-to-treat principle.
Treatment phase
Study outcomes will be described as means and SD. Transformations may be required when the distributions are non-normal. Enrolment characteristics will be described for the whole trial and separately by type of cancer. The primary outcome of MSAS total score is continuous and will be assessed in a repeated-measures analysis using mixed models. Hence, the analysis will test the difference between ASyMS and standard care groups in the change in symptoms between enrolment and repeated follow-up. The primary hypothesis will be tested through the regression parameter for the ASyMS versus standard care groups, adjusting for enrolment MSAS as well as stratified by type of cancer and country. Adjustments will be made for length of treatment, age, gender, as well as symptom prevalence and severity at enrolment. The prespecified subgroup analyses by type of cancer, country, age, gender and symptom prevalence and severity will be assessed by fitting trial arm by subgroup interaction parameters.
Should the active recruitment period of the RCT over run, it is acknowledged that this may affect the number of patients with data at all time points at the designated end of the follow-up. During the follow-up period, a separate analysis will be performed to indicate how many patients would be required by the end of the follow-up period.
Follow-up phase
Two mixed-models analyses will be carried out. First, the repeated measures of outcomes in the extended follow-up will be added to those already obtained from the active CTX period. This analysis will be a longer-term follow-up of the active CTX period and has the advantage of further repeated measures adding power to the comparison. It will test whether any effect seen after the trial is sustained for up to a year. Post-CTX management is highly individualised. Therefore, groups of patients are expected to receive different maintenance treatment based on cancer diagnosis and disease characteristics and different models of follow-up (eg, traditional, open access, risk stratified). Therefore, additional subgroup analyses will be performed and adjustments made for these differing characteristics in the modelling.
Second, a separate analysis will take baseline as the end of intervention and analyse the repeated measures of the outcomes up to 12 months. This analysis will be an observational cohort analysis of the postintervention stage and will require that more confounding factors be taken into account. The analyses will use mixed models as in the active intervention period. The extent of missing data in the outcomes will be explored and the reasons for missing data noted.
Monitoring
The Surrey CTU has developed a monitoring plan that will record all of the information regarding monitoring. Each clinical site will be responsible for performing their monitoring. In addition, a Data Monitoring Committee (DMC) was established. The role of the DMC is to review accumulating study data related to the safety and efficacy of the study intervention and to ensure continued scientific validity and merit of the study.
Safety reporting
Any study-related incidents will be reported to the national competent authority in each country. The local ethics committee that granted ethical approval in each country will be informed.
Trial status
The RCT is now open and actively recruiting patients.
Acknowledgments
The authors are grateful to the clinicians at the clinical sites who are integral to this study (patient recruitment, alert handling, symptom assessment and management and data collection) (see online supplementary appendix 1).
References
Footnotes
Contributors NK and RM developed the initial trial concept. NK, RM, LM, GK and MM devised the study design, drafted the protocol and contributed to critical revisions of the manuscript. CM contributed to the study design and critical revisions of the protocol and the manuscript. PF drafted the manuscript and contributed to revisions of the protocol. EF contributed to revisions of the protocol and critical revisions of the manuscript. ER, JA, PM, JH contributed to the study design and critical revisions of the protocol. EP, SK, AG, GVB, KA and AF contributed to critical revisions of the protocol. AB contributed to revisions of the manuscript. PD carried out the sample size calculations. PD wrote the statistical analysis plan.
RM is the Chief Investigator and takes overall responsibility for all aspects of trial design, the protocol and the trial conduct. All authors have read and approved this manuscript.
Funding The eSMART project has received funding from the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 602289.
Competing interests None declared.
Ethics approval The eSMART study has received ethical approval from NHS Lothian, South East Scotland Research Ethics Committee (14/SS/1062). Ethics approval has also been received from all of the following: St. Vincent’s Healthcare Group Ethics and Medical Research Committee, Ireland; St James’s Hospital/Adelaide, Meath incorporating National Children’s Hospital Research Ethics Committee, Ireland (2015 List 27(5)); REK Regional Committees for Medical and Health Research Ethics, Norway (REC South East 2015/408 eSmartstudien). Hellenic Data Protection Authority (no.: Γ/ΕΞ/2630/07-05-2015); National and Kapodistrian University of Athens, Faculty of Nursing, Ethics and Research Committee (no.: 138, 15/1/2015), Agioi Anargiri Cancer Hospital Ethics and Research Committee (no.: 267, 13-3-2015) (no.: 839, 15-9-2015), Metropolitan Hospital Ethics and Research Committee (19/5/15) (4/9/15), Air Force General Hospital Ethics and Research Committee (no.: 6048, 3/4/15, 23/10/2015), Ethics Committee of the Medical University of Vienna (1165/2015).
Provenance and peer review Not commissioned; externally peer reviewed.