Article Text

Abstract
Objectives We examined major issues associated with sharing of individual clinical trial data and developed a consensus document on providing access to individual participant data from clinical trials, using a broad interdisciplinary approach.
Design and methods This was a consensus-building process among the members of a multistakeholder task force, involving a wide range of experts (researchers, patient representatives, methodologists, information technology experts, and representatives from funders, infrastructures and standards development organisations). An independent facilitator supported the process using the nominal group technique. The consensus was reached in a series of three workshops held over 1 year, supported by exchange of documents and teleconferences within focused subgroups when needed. This work was set within the Horizon 2020-funded project CORBEL (Coordinated Research Infrastructures Building Enduring Life-science Services) and coordinated by the European Clinical Research Infrastructure Network. Thus, the focus was on non-commercial trials and the perspective mainly European.
Outcome We developed principles and practical recommendations on how to share data from clinical trials.
Results The task force reached consensus on 10 principles and 50 recommendations, representing the fundamental requirements of any framework used for the sharing of clinical trials data. The document covers the following main areas: making data sharing a reality (eg, cultural change, academic incentives, funding), consent for data sharing, protection of trial participants (eg, de-identification), data standards, rights, types and management of access (eg, data request and access models), data management and repositories, discoverability, and metadata.
Conclusions The adoption of the recommendations in this document would help to promote and support data sharing and reuse among researchers, adequately inform trial participants and protect their rights, and provide effective and efficient systems for preparing, storing and accessing data. The recommendations now need to be implemented and tested in practice. Further work needs to be done to integrate these proposals with those from other geographical areas and other academic domains.
- clinical trials
- individual participant data
- data sharing
- consensus conference
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
An effective and formal consensus-building process among a large group of very experienced researchers and others involved in clinical trials.
A unique perspective: Europe-wide, non-commercial, with a focus on the particular needs of researchers.
A large number of practical recommendations set against an overarching framework of principles.
The recommendations now need to be implemented and tested in practice, and feasibility and usability should be explored.
The exercise is largely based on experience and opinions, and members of the task force may be not fully representative of the research community.
Introduction
Background
In recent years, several major organisations have called for increased sharing of the data generated by publicly funded research, including the Organisation for Economic Co-operation and Development,2 the European Commission,3 the National Institutes of Health (NIH) in the USA4 and the G8 science ministers.5 This trend reflects the growing recognition that ‘Publicly funded research data are a public good, produced in the public interest, which should be made openly available with as few restrictions as possible in a timely and responsible manner’.6
Data from clinical research are not exempt from this call, even though concerns over participant privacy mean that such data often need to be specially prepared (eg, de-identified) before they can be shared. Given the key evidential role that clinical trials play in determining evidence-based medicine and evidence-based public health policies, sharing this type of data is seen as particularly important. Indeed, it has been argued that clinical trial data should be shared and treated as a public good whoever generates it, that is, whether they are created by publicly funded or commercial research.7
Sharing data from clinical research can be justified on scientific, economic and ethical grounds.8 Scientifically, sharing makes it possible to compare or combine the data from different studies, and to more easily aggregate it for meta-analysis. It allows conclusions to be re-examined and verified or, occasionally, corrected, and it can allow new hypotheses to be tested. Sharing can therefore increase data validity, but it also squeezes more value from the original research investment, as well as helps to avoid unnecessary repetition of studies. The economic advantages of data reuse are one reason why governmental and intergovernmental agencies, as well as major research funders (eg, the Gates Foundation9 and the Wellcome Trust10), support data sharing.
Ethically, data sharing provides a better way to honour the generosity of clinical trial participants, because it increases the utility of the data they provide and thus the value of their contribution. It is also argued that, if access to health and healthcare is a basic human right, access to data that can improve health is similarly a fundamental right,11 and those involved in research, and its governance and funding, have an obligation to their fellow citizens to respect and promote that right.12
The rapid acceptance of the idea of sharing clinical trial data was summarised in 2016 by Vickers,13 who was able to claim a ‘tectonic shift in attitudes’ over 10 years. Turning the idea of data sharing into a reality, so that it becomes ‘an unquestioned norm’ (to borrow Vickers’ phrase), certainly requires a change in attitudes, but there also needs to be an appropriate policy environment, adequate resourcing, clarity about the roles and responsibilities of different stakeholders, specific objectives and indicators to measure progress, and an available digital infrastructure.
Origin of this document
The document has been prepared in the context of a specific working task of the European Union (EU) CORBEL (Coordinated Research Infrastructures Building Enduring Life-science Services) project (www.corbel-project.eu). CORBEL is designed to establish a collaborative and sustained framework of shared services across 11 participating European, European Strategy Forum on Research Infrastructures (ESFRI) biological and medical research infrastructures, to better support biomedical research in Europe and accelerate its translation into medical care.
One of the objectives of this working task is to develop procedures to provide the scientific community with access, on request, to the individual participant data (IPD) from previous clinical trials for reanalyses, secondary analyses and meta-analyses. This activity is led by the European Clinical Research Infrastructure Network (ECRIN-ERIC), an ESFRI research infrastructure that provides guidance, consulting and operations management for multinational clinical trials on a not-for-profit basis (www.ecrin.org). ECRIN already requests that the investigators it supports commit to make anonymised IPD data sets available to the scientific community on request.
To be clear, throughout this document we use IPD to refer to all of the participant data available from a trial, and not just the data supporting the conclusions of a specific published paper. Such data will therefore normally be the data sets used for the various analyses, after appropriate de-identification and pseudonymisation or anonymisation measures have been applied. The goal is to develop a framework in which, ultimately, all of the participant-level data from any trial become available to those who can demonstrate they can make appropriate use of them.
Various other organisations have also addressed this task in recent years and developed generic principles as well as practical recommendations for implementation of data sharing. Usually, these documents are embedded in a geographical/national context (eg, the Institute of Medicine report in the USA,14 the Nordic Trial Alliance Working Group on Transparency and Registration for the Nordic countries,15 the good practice principles for sharing IPD from publicly funded trials by Medical Research Council (UK) (MRC), UK Clinical Research Consortium (UKCRC), Cancer Research UK (CRUK) and Wellcome in the UK,16 17 or the guide to publishing and sharing sensitive data for Australia18).
Other groups have examined clinical research data sharing within a much wider context, such as the principles of data management and sharing within European research infrastructures developed by BioMed Bridges.19 Conversely, other initiatives have been centred on a specific stakeholder group, such as the pharmaceutical industry (eg, the principles for responsible clinical trial data sharing produced by Pharmaceutical Research and Manufacturers of America (PHRMA) and European Federation of Pharmaceutical Industries and Associations (EFPIA)20), or on specific subsets of clinical trial data (eg, the 2016 International Committee of Medical Journal Editors (ICMJE) proposal was focused on the data underlying the results presented in an individual journal article21).
These and other documents were taken into consideration in our consensus exercise and, as a consequence, in this report. Nevertheless, we believe that in this report we have been able to bring a broader international perspective on data sharing in clinical trials, reflecting the professional and geographical diversity of our expert group. We have also tried to examine all stages of the data sharing ‘life cycle’, including the following:
Supporting trialists, for example, in planning for data sharing and in preparing data.
Suggesting the best policies and practice for data and metadata storage.
Promoting data discovery and discussing data access mechanisms and agreements.
The intention was to examine all the major issues associated with sharing IPD and trial documents, using a broad, multidisciplinary approach. Inevitably, however, certain perspectives have been emphasised, as described below.
The perspectives of this document
Trials or studies?
The remit of the task group was to look at data sharing from clinical trials, rather than clinical studies in general (the latter term including trials and non-interventional studies, both prospective and retrospective, including epidemiological and registry studies—see the glossary for formal definitions in online supplementary 2). Although we have largely kept to that restriction, it should be acknowledged that many, probably most, of the principles and recommendations have relevance to clinical studies in general. That is sometimes reflected in the text, when ‘study’ is used rather than ‘trial’, but it is stressed that the formal scope of this document remains clinical trials.
Supplementary file 2
Non-commercial trials
The emphasis of the project was on data sharing from non-commercial trials, partly because most of the expert group members have a background in non-commercial research. In addition, many of the existing non-commercial IPD sharing initiatives were perceived as having a limited scope, for example involving only specific collaborative trial groups or disease-specific activities. The task force was therefore keen to develop more generally applicable policies and guidance. Solutions developed in collaboration with the pharmaceutical companies (eg, Yale University Open Data Access (YODA)22 and Clinical Study Data Request (CSDR23)) may be applicable to the academic world, but so far this has not been tested. CORBEL wants to develop procedures and tools for the whole scientific community, while remaining complementary to existing initiatives. We believe that most if not all of the recommendations presented here are also applicable to IPD generated in the commercial sector. It should be noted that non-commercial clinical trials make up approximately 40% of the trials conducted in Europe.24 25
A European origin
The CORBEL project is funded by the EU and has a clear European perspective. Although several members of our working group represent institutions from non-European countries (USA, Canada, Australia, observers from Japan) and we feel strongly that most of the recommendations have global scope, it is true that our discussions often referenced a European context, for instance when discussing personal data protection legislation. As many current initiatives about data sharing have a US base (eg, the Institute of Medicine,16 the Multi-regional Clinical Trial Centre (Harvard University) (MRCT) Center Vivli project,26 and most of the ICMJE members), it could be argued that a European perspective is required, especially given the potential differences in legal frameworks as they relate to data sharing. It is also timely, given that the European Commission is pushing strongly for open access to scientific information, including supporting the development of a new European Open Science Cloud (EOSC) with major investment from the European Horizon 2020 research programme.27 It is expected that sensitive data from clinical trials will constitute a major use case within this initiative. If successfully implemented, the EOSC could therefore provide a suitable infrastructure to host and share clinical trial data and documents.
The perspective of the researcher
The emphasis throughout has been on the perspective of clinical researchers, considered both as data generators and as data requesters/(re)users.
To be clear, by ‘data generators’ we mean the trialists and other study personnel that conceive of the study, and then plan, manage, monitor, analyse and publish it. This requires a complex set of intellectual and organisational skills, and we do not wish to suggest that a trial can be reduced to mere ‘data generation’, or that the term ‘data generators’ is used in any way in a derogatory sense. It is simply that, in this context, the term usefully emphasises the role of the trial as the data generation phase, and the role of the trialists as the designers and initial creators of the data set.
Other actors (funders, publishers, infrastructure providers) are all of course vitally important, but the main target group for this document are researchers themselves. We hope that this document will raise awareness of IPD sharing among data generators and also show how, with suitable policies and tools, concerns about data sharing can be reduced.
Because publications and citations are of utmost importance in the academic world, the project also aims to promote data as a legitimate, citable product of research, and to ensure that making data available for sharing is recognised and rewarded. We have also tried to examine the needs of those searching for data and trial documents, emphasising the importance of discoverability and the need for transparent but relatively simple mechanisms for requesting and gaining access.
The aim of this document is to help turn the sharing of data from clinical research, in particular from clinical trials, from an aspiration to accepted practice. It does so by first proposing a set of overarching principles that we think should guide the practice of data sharing, and then examining the policy and practical issues associated with each and making a series of recommendations.
Methods
This consensus exercise was carried out in a series of three workshops held over 12 months (March and October 2016, March 2017), supported by exchange of documents and teleconferences within focused subgroups when needed. Successive drafts of the report were circulated before each workshop, with final versions being circulated for comments, suggestions and agreement after the third workshop. The applied methodology was based on the nominal group technique to ensure that all participants had a chance to formulate and contribute their opinions and to vote on the proposals.
The nominal group process28 29 is a strict, formal procedure to facilitate innovation and creativity while still achieving consensus. It consists of the following steps:
Proposal of a text by a core group.
Comment from each group member.
Collection of comments by the moderator.
Collapsing of similar comments.
Prioritisation of discussion points.
Discussion of all comments.
Voting on each discussion point.
Rewriting of text by core group according to voting results.
Revision of new text by starting again at step (1) until consensus is reached.
The iteration process of step (9) was implemented by starting with a new revised text version at each workshop.
ECRIN established a core group responsible for the management of the consensus exercise and preparation of the consensus document. The group included experts in multinational clinical trials, trial methodology and transparency, trial management services, information technology (IT) tools, and legal issues. The core group’s responsibilities were to establish the multistakeholder task force, draft intermediate versions of this report, organise and manage the consensus workshops, coordinate the subgroups, and release the final version of the report.
Given the complexity of the issues around sharing and reusing data from clinical trials, any attempt to develop principles and procedures requires the involvement of a wide range of stakeholders to represent the different groups generating, managing and using IPD. It was also important to ensure that a range of scientific, technical and legal expertise was present, and that different geographical regions were represented in the discussion. A multistakeholder task force was therefore assembled including researchers, patient representatives, methodologists, information technology (IT) experts, and representatives from funders, infrastructures and standards development organisations, as well as the core group members, to evolve the consensus reported in this document.
Consensus building among the task force was carried out with the support of an independent facilitator, who cochaired the meetings and provided guidance on the consensus process and how to handle and report written feedback on the intermediate versions of the report. Online supplementary appendix 1 lists the full membership of the core group and multistakeholder task force.
During the first workshop, the task force agreed on the establishment of two subgroups to provide insights to the consensus exercise. The first subgroup worked on terminology, to clarify the main terms used in the project based on legal definitions, regulations and standards. The output of this subgroup is the glossary of standardised terms and definitions reported in online supplementary appendix 2. The second subgroup worked on an environmental scan of the existing data sharing repositories and other initiatives relevant for sharing of IPD, to describe current provision and highlight possible missing features or functions. The output of this subgroup will be reported in a separate publication.
Results
Ten principles emerged from the consensus process, representing what the task force saw as the fundamental requirements for any framework for the sharing and reuse of clinical trials data. They are listed in box.
Principles of data sharing in clinical trials (P, principle)
P1: The provision of individual participant data should be promoted, incentivised and resourced so that it becomes the norm in clinical research. Plans for data sharing should be described prospectively, and be part of study development from the earliest stages.
P2: Individual participant data sharing should be based on explicit broad consent by trial participants (or if applicable by their legal representatives) to the sharing and reuse of their data for scientific purposes.
P3: Individual participant data made available for sharing should be prepared for that purpose, with de-identification of data sets to minimise the risk of reidentification. The de-identification steps that are applied should be recorded.
P4: To promote interoperability and retain meaning within interpretation and analysis, shared data should, as far as possible, be structured, described and formatted using widely recognised data and metadata standards.
P5: Access to individual participant data and trial documents should be as open as possible and as closed as necessary, to protect participant privacy and reduce the risk of data misuse.
P6: In the context of managed access, any citizen or group that has both a reasonable scientific question and the expertise to answer that question should be able to request access to individual participant data and trial documents.
P7: The processing of data access requests should be explicit, reproducible and transparent, but, so far as possible, should minimise the additional bureaucratic burden on all concerned.
P8: Besides the individual participant data sets, other clinical trial data objects should be made available for sharing (eg, protocols, clinical study reports, statistical analysis plans, blank consent forms) to allow a full understanding of any data set.
P9: Data and trial documents made available for sharing should be transferred to a suitable data repository to help ensure that the data objects are properly prepared, are available in the longer term, are stored securely and are subject to rigorous governance.
P10: Any data set or document made available for sharing should be associated with concise, publicly available and consistently structured discovery metadata, describing not just the data object itself but also how it can be accessed. This is to maximise its discoverability by both humans and machines.
The task force also agreed 50 more detailed recommendations, grouped around seven major topics, each associated with one or more principles, as shown in figure 1.

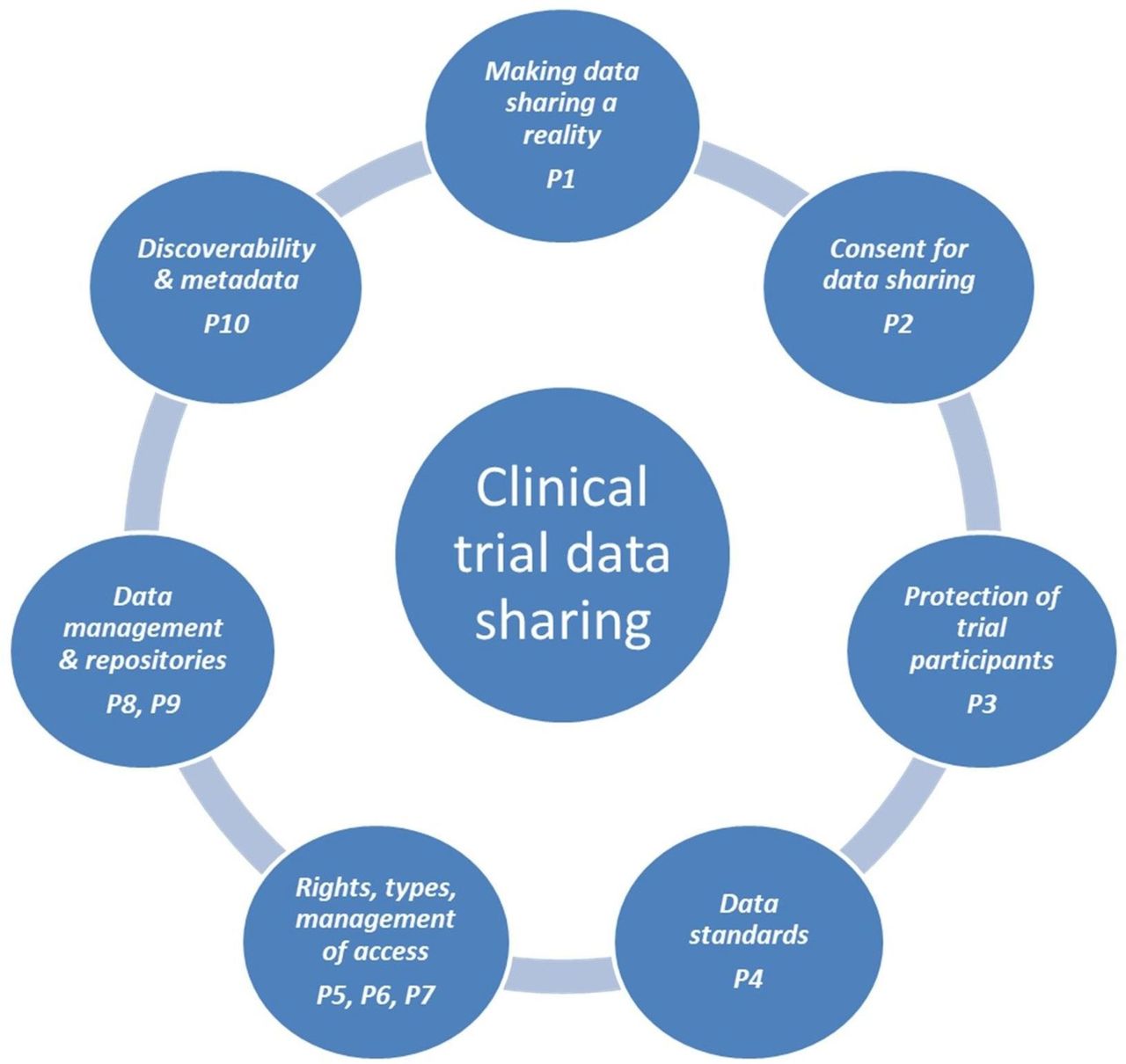{kind=link}
Major aspects of sharing and reuse of data from clinical trials. P, principle.
These seven topics have been used to structure the lists of recommendations that follow. Each section also includes explanatory text for both principles and recommendations.
Making data sharing a reality
P1: The provision of IPD should be promoted, incentivised and resourced so that it becomes the norm in clinical research. Plans for data sharing should be described prospectively, and be part of study development from the earliest stages.
There is now widespread acceptance of the need for greater sharing of IPD, but much of the pressure for this has been ‘top-down’—it has come from funding organisations, professional bodies and journal editors (although some ‘bottom-up’ sharing activity also exists, eg, within collaborative research groups). Some researchers retain misgivings, for instance about the resources required to support data preparation, or potential misinterpretation of their data, or a possible reduction in the number of papers they will be able to generate from the data themselves. These fears need to be recognised and mitigated by appropriate resourcing, policies and systems, including changes to the way research activity is recognised and rewarded. Such developments are necessary if making IPD and related study material available is ever to be seen as a normal, integral part of clinical research, accepted as such by the researchers themselves.
To help make that happen, researchers will need support, to ensure that data sharing is considered from the very beginning of study planning. Trying to organise safe and effective data sharing retrospectively, especially if appropriate consent and resourcing have not been obtained, will often be difficult, complex and expensive, and many non-commercial researchers would have great difficulty in justifying the additional input required. Making provision for future data sharing a standard component of study design is therefore essential.
All stakeholders involved in clinical research (eg, funders, patients’ groups, researchers, academia, professional groups, industry, editors, and regulatory and ethics authorities) should support sharing of IPD and study documents as a normal part of good practice.
Most of the major stakeholders in clinical research do recognise the importance of sharing IPD and trial documents, and many have made public statements to that effect. But these changes in attitude have to be turned into practical measures of support. No single group can be held responsible as the main drivers of data sharing, and responsibility (and resourcing) needs to be shared—each stakeholder group will therefore have to evolve their own role within this developing field.
For example, actions taken by the European Medicines Agency in Europe,30 by the US Congress with the 21st Century Cures Act in the USA31 and by the WHO in the context of public health emergencies32 represent policy changes with respect to data sharing at national and international levels, but the full implications of such changes will often need to be clarified. Public funding agencies (eg, NIH in the USA) and funding charities (eg, Wellcome Trust, Bill and Melinda Gates Foundation) increasingly require that the studies they fund include data management and sharing strategies, but the practical limits to the financial support for data sharing from funders need to be explored. Biomedical journals, as exemplified by the ICMJE, are developing data sharing policies that will oblige authors to make the curated data and metadata supporting their findings available,14 although the timing of such availability is the topic of debate. International organisations that consider ethics in clinical research, for example the World Medical Association, have also issued statements about data reuse,33 and stakeholders will need to develop a consistent interpretation of such principles.
The promotion of a culture of data sharing and reuse will therefore require an ongoing dialogue between all parties, parallel to the efforts aiming to encourage and monitor data sharing. Short-term projects such as CORBEL can play an important role in stimulating that dialogue, but more permanent infrastructure organisations, such as ECRIN, Biobanking and BioMolecular Resources Research Infrastructure (BBMRI) and European Institute for Innovation through Health Data (i~HD), are likely to have a key role in orchestrating such discussions in the longer term.
Any data sharing model should be based on the concept of data ‘stewardship’ rather than data ‘ownership’.
The data generated in the context of clinical research activities should be seen as a public good—that is, one that is common to humanity as a whole. We believe that is the only way to properly recognise the value of the data and the generosity of the study participants who provided them. Although the researchers who generate the data may have the greatest stake in their use, they should not perceive it as their ‘private property’. In fact (and despite the various practical issues that we discuss throughout this document) they have a responsibility to ensure the data are discoverable by others and accompanied by sufficient metadata for them to be found easily, understood in context and used appropriately. Commonly the term ‘stewardship of research data’ is used to summarise this approach, which includes providing useful accessibility, annotation, curation and preservation of the data.34
We recognise that there are not, currently, formal definitions of ‘stewardship’ and ‘ownership’ of data that are universally accepted. There are specific uses of both terms, linked to debates about (for instance) contracts, copyright and intellectual property rights, but within the consensus conferences we wanted to keep clear of these more legal and technical issues. We wished to stress instead that the concept of stewardship—as described above—should be the default assumption for IPD sharing, to be used when developing a policy framework and by individual researchers when considering their own data sharing strategies.
Academic and societal rewards for data sharing should be implemented so that making data available for data sharing is seen by researchers as an opportunity. Such incentives might include recognition in the assessment of academic careers or grant proposals.
For researchers, planning, performing and analysing a clinical trial is a difficult, resource-intensive and lengthy exercise. In the academic world, reputation and career are mainly based on scientific presentations and publication of research results. Data sharing may be highly desirable from a societal or ethical viewpoint but, up to now, the academic benefit for the data generators has been limited, although some analyses have reported that the citation rate of a publication is higher when its data are made publicly available.35
To help convince data generators to share their data, stronger incentives are necessary. The reuse of data sets generated by researchers should be valued in the assessment of academic careers, including for promotion, as part of a more comprehensive evaluation of the professional work of trialists. Shared data sets therefore need to become an acceptable academic coinage. Agreed mechanisms for including data sharing in academic career assessment are not yet available, but a variety of detailed proposals have been made and will need to be tested in practice.36 37 The evaluation of funding applications should also take into account the applicant’s past record of making IPD data available for sharing, and the subsequent level of reuse of that data.
Clinical trial data sets should be considered legitimate, citable products of research. To support citability they must each have a persistent and globally recognised identifier.
Persistent identifiers, such as the already widely used digital object identifier (DOI), should be applied to data sets to improve discoverability and to allow correct citation. The issue of data citation is currently being intensively addressed,37–40 and it is hoped that widely accepted procedures for data citation will evolve in the very near future. For example, the Force11 Data Citation Synthesis Group has published a Joint Declaration of Data Citation Principles,41 which has been endorsed by 94 repositories, publishers and scholarly organisations, including DataCite, Committee on Data (of the International Council for Science) (CODATA) and the Nature Publishing Group.42 In addition, several organisations and publishers have introduced metrical instruments for data citation.43 44 Identifier, citation and citation metric schemes are an essential prerequisite for the broad acceptance and implementation of data sharing.
A potential problem in assigning identifiers is that different versions of data sets and documents may be available. For instance, trial protocols are often amended and consequently assigned different version numbers, or a long-running study might generate additional follow-up data. Even data generated at the same time may exist in different forms, for instance trial analysis data versus the same, partly uncoded, data set, as originally collected on (e)CRFs. Versioning is a problem common to many types of data storage, and various technical approaches have been proposed—the simplest being distinct DOIs for different versions, but with the linkage between versions retained explicitly in other metadata elements. The key point we make here is that a generally applied versioning scheme would be a necessary part of any overall approach to assigning identifiers to trial data sets and documents.
Stakeholders involved in clinical research need to develop fair and sustainable financial models for data sharing, to ensure the long-term resourcing of data preparation and storage as well as the request and sharing process.
The costs of preparing data for secondary use, its subsequent maintenance in repositories and the request and access processes all need to be adequately funded. Inclusion of initial preparation costs in funding applications is probably the most obvious option, but different mechanisms for sustainable funding of data sharing need to be explored. We believe that charging fees for access to data should be avoided wherever possible, as it could discourage applications for access, especially from academic researchers and from low-income or middle-income countries. We accept, however, that there may be situations (eg, for legacy trials) where some of the costs of preparing data for sharing may need to be met by the secondary users, or it will be difficult to make the data available. Irrespective of the business model adopted, the final goal must be to encourage data sharing and reuse.
Long-term storage and access costs are not easily predictable and thus not easily linked to initial funding.
Possible sources of support include core/structural funding, hosting organisations or private contracting, data deposition fees, access charges, or research and development (R&D) project funding.45 The discussion on sustainable business models for data infrastructures is ongoing, and it is difficult to identify a preferred model. A particular problem is that while many established national and international data repositories have core streams of income from research funders, these sources of income are usually short-term and may be vulnerable to change in priorities or in responsibilities. The Organisation for Economic Co-operation and Development Global Science Forum is working with partners on two projects related to Open Data for Science, one on the sustainable business models for data repositories and a second on international coordination of data infrastructures.46
To ensure more effective and widespread sharing of IPD and other data objects, organisations should be encouraged to revise their policies to allow wider data reuse.
Sometimes local policies, implemented by research institutes and universities, may restrict data sharing possibilities for the data generators. These policies can derive from a variety of historical beliefs, including a general distrust of data reuse, perhaps negative prior experiences, worries about academic competition, and concern over ownership and copyright issues.47 But such beliefs are incompatible with the new global attitudes towards data sharing and reuse, and institutional policies should be reviewed to try and ensure that such barriers are removed.
Data sharing should be prospectively planned, described within a designated section of the trial protocol and summarised in the relevant section of the trial registration record.
To ensure data sharing is considered from the beginning of a trial, it should be included within the trial protocol. This is also suggested by other initiatives, for example in ref 17, and mentioned as a standard item in protocols for interventional trials by the standard protocol items: recommendations for interventional trials (SPIRIT) guideline, under ‘Dissemination policy’:
‘The protocol should indicate whether the trial protocol, full study report, anonymised participant-level dataset, and statistical code for generating the results will be made publicly available; and if so, describe the timeframe and any other conditions for access’.48
The description of how IPD will become accessible should therefore be much more than a vague statement of intent. It would be useful also to include this information in the trial’s registry entry. WHO-adopted registries, such as ClinicalTrials.gov and international standard randomised controlled trial number (trial registry) (ISRCTN), have started to include basic information on publication and dissemination plans and availability of IPD. Following an International Clinical Trials Registry Platform (ICTRP) registry network meeting in 2017, it is expected that new data elements will in time be collected by more registries and displayed through the WHO portal.
All the trial documents (eg, participants’ information leaflet, contracts, consent forms, ethical submission documents) should be written taking into account the planned data sharing strategy.
As a consequence of planning data sharing from trial inception, other documents can be written to take that data sharing into account. Participant information leaflets should summarise the plans for data sharing, including the use of external repositories, and consent forms should include the relevant requests for consent (see following section on consent). The data management plan, as well as other documents submitted for regulatory and ethical review, should refer to the planned data sharing strategy and related actions. It is not yet the case that ethical approval is contingent on planned data sharing, but we suggest that data sharing plans should be open to ethical scrutiny. Ethics committees could play an important role in facilitating responsible data sharing, for instance by assessing plans and ensuring that appropriate information and consent forms are used.49
To help support the implementation of data sharing within trial planning, services providing support and storing example documents should be provided.
As a relatively new activity, planning for data sharing may be difficult for many researchers. Having example documents and templates (eg, of consent forms and protocol sections) may therefore be a useful practical step in promoting data sharing as a normal trial activity. The provision of advisory services that can make such material available may also be useful. There is no suggestion that each institution should develop its own service, but an organisation acting at national or supranational level could usefully gather and disseminate examples of good practice.
The time for making IPD data and documents available for reuse will vary, but times should be monitored and investigated to identify and normalise reasonable expectations.
It is difficult to make a statement that is too prescriptive about the timing of ‘release’ of full IPD data sets for reuse. Other initiatives have attempted to define timelines: for example, the Institute of Medicine report suggested that clinical trial data that will not be part of a regulatory application be made available for sharing no later than 18 months after study completion.15 The ICMJE originally suggested that data underlying the results presented in a journal paper be shared no more than 6 months after publication,21 although more recently, perhaps mindful of some of the practical issues we discuss in this paper, they have provided much more flexible guidance.50
We believe the goal should be to make trial data and documents available in a timely manner. But the exact time will depend—for instance—on the possibility and timing of publications by the primary investigators, the complexity of the study and any associated substudies, the nature of the documents or data, the amount of analysis and preparation the data might require, and the access regime under which it is planned to make it available.
There is an expectation that most trial documents (other than those describing the aggregate results, such as a clinical study report), could and should be released soon after the end of data collection. For the IPD data sets, however, we believe investigators should be confident that they have completed their own planned authorship activity before making the whole of the IPD data set available. We think it reasonable, however, to expect de-identified data supporting a specific published paper to be available relatively quickly, normally within 1 year of that paper’s publication. In addition, although different portions of the data set derived from a trial may be released at different times, we believe (along with the ICMJE50) that investigators should clearly indicate when they anticipate all the data will be released. In other words, the data sharing plan should include a time limit, available for inspection at the beginning of the study and for comparison, with actual data release, after the study has finished.
It will be important in the future to monitor when IPD is made available, and the access regimes that are used, comparing the reality with the data sharing plans originally proposed. Such monitoring will inevitably require support and funding from research infrastructures, but it will be necessary to identify not just the volume, nature and timing of data reuse, but also the technical, attitudinal and financial barriers that might impede it. That will facilitate both targeted input to minimise those barriers, and lead to a better, shared understanding of what are reasonable expectations for the timing of data release.
Consent for data sharing
P2: IPD sharing should be based on explicit broad consent by trial participants (or if applicable by their legal representatives) to the sharing and reuse of their data for scientific purposes.
The process of informing trial participants about possible sharing of their data, and then gaining their explicit consent to it, is of fundamental importance, and is normally a prerequisite for the sharing of pseudonymised data (ie, data that have been de-identified but that can still be linked back to individuals using additional but separately stored material—see the glossary in onlinesupplementary appendix 2 for further details).
Data sharing activities that are an integral part of a trial (eg, data transfer between collaborating groups) can be anticipated and described in the information given to participants, and so can be included within the informed consent for trial participation. But the nature, purpose and destination of IPD sharing that may occur after the trial completes are impossible to predict. By definition, therefore, any consent for this secondary use of data cannot be fully ‘informed’. Instead what should be sought from the participant is a ‘broad’ consent to their data being shared, with the caveat that it should be shared only for scientific purposes.
It is worth noting that the European General Data Protection Regulation’s (GDPR)1 requirement, that the data subject be fully informed about the purpose of data processing at the time of data collection, is less strict when it comes to scientific research. For instance, Recital 33 of the GDPR suggests that:
It is often not possible to fully identify the purpose of personal data processing for scientific research purposes at the time of data collection. Therefore, data subjects should be allowed to give their consent to certain areas of scientific research when in keeping with recognised ethical standards for scientific research. […]
The EU clinical trial regulation 536/2014 also refers to reuse of data from clinical trials for future scientific research, underlining the importance of the consent to use data outside the protocol of the clinical trial, the right to withdraw that consent at any time, and mechanisms to review that secondary analyses are appropriate and ethical (paragraph 29 of the preamble).51
Broad consent should still be given with as much information as is practicable, for instance about the reasons for data sharing (in general, not as it might relate to their own data) and the nature of any preparation of the data prior to them being shared (for instance a statement saying that it will be de-identified). Like all consent, to be meaningful it must also be given without coercion, however unintended that coercion might be. In particular, the consent should be explicit and clearly separate from any other consent. It cannot be implied by the consent to participate in the trial, because it is a separate activity and not part of that trial (although as explored in the discussion section, we accept that not everyone holds this view). Nor can consent to data sharing be used as an inclusion criterion for the trial, as this implies coercion.
It has been argued that if participants need to provide separate consent for data sharing, there is a danger that any shared data set will differ from that used in the original analysis, that is, that participants who do not agree on sharing their data are systematically different from those who agree, producing a bias in the population under study. Because of this it is argued that consent to data sharing should be assumed unless an ‘opt-out’ option is exercised. One difficulty with the ‘opt-out’ approach is that this is not a valid concept in many EU countries, but the more fundamental problem is that it is not a form of explicit consent. In fact, it would create only an implicit consent, and we believe that would form an inadequate basis, legally and ethically, for later data sharing actions.
Gaining consent to secondary use of data should become a standard procedure, to provide legitimate sharing of data collected during clinical trials.
This recommendation follows as an obvious consequence of the principle above. Gaining explicit broad consent is the only simple way to avoid the legal complexities of attempting to share data where such consent does not exist. Even though, in some jurisdictions, explicit consent for the secondary use of fully anonymised clinical trial data may not be legally necessary, there are problems with what ‘fully anonymised’ might mean in practice. In addition, the legal context continues to evolve, for instance with the introduction of the GDPR1 in Europe, and future national modifications and judicial interpretations of that regulation, and it is difficult to predict possible limitations on the use of data without consent. Beyond this pragmatic requirement for gaining consent, there is also an ethical imperative to be open and transparent with participants about the possible use of their data, which should make seeking explicit consent for data sharing mandatory.
Normally, the explicit consent for data sharing should be provided at the same time of the informed consent for the clinical trial participation.
Although separate, the consent to IPD sharing should normally be obtained at the same time as the consent for participation in the trial. This makes the whole process more practical and less of a burden for both investigators and participants. There will be some circumstances when this is difficult (eg, emergency care situations), and the consent to secondary use of data may therefore necessitate a separate consent event.
The consent for secondary use of IPD should be as broad as possible.
The broad consent given should allow the future scientific use of the data. Restricting future secondary use to research in particular disease areas or types of research, for example, should be avoided, because it will be impossible to predict the source of requests for data access and how they might be categorised. The concept of broad consent comes from the field of biospecimens and biobanks, where it is generally accepted from an ethical perspective, especially when there is a process of oversight and approval of future research activities.52 We therefore recommend a broad consent for ‘data sharing for scientific purposes’, which explicitly excludes any other, for example, for insurance or forensic purposes.
An appropriate consent process for secondary use of data should ensure the following:
The reasons for asking about data sharing, and the general benefits of data sharing in clinical research, are made clear to the trial participant.
Although it is envisaged that most trial participants will willingly consent to data sharing, it is still important that potential trial participants are informed about the general benefits of such sharing for science and medical practice. This information is likely to be part of the patient information sheets.
The nature of data preparation, storage and access is explained to the trial participant, so far as they are known at the time the patient documents are produced.
It will also be important to describe, in broad terms, how and where the data will be stored, and how confidentiality will be maintained (eg, by de-identification measures). Even though consent for data sharing cannot be fully informed, because the nature, purpose and destination of data sharing that may occur after the trial completes are impossible to anticipate, efforts should still be made to describe the measures that will be used to protect participant privacy, the type of requests that will be considered and the scrutiny to which they will be subjected, and so on. In other words, the consent should be as informed as possible. Obviously, this requires at least the outlines of a data sharing strategy to be in place from the outset of the trial.
The information provided should be clear and concise, and couched in vocabulary understood by the trial participants (or if applicable their legal representatives).Further research is needed to identify appropriate ways of presenting this information to the participant, and good practise needs to be defined and implemented.
The explicit consent for data sharing should be reflected in the layout of the consent forms.
A request for consent to secondary use of data must be clearly distinguishable from any other matters in the informed consent document. This does not mean, however, that separate consent forms or documentation are required to handle data sharing—the different signature sections can be integrated into one document, and it would normally be easier to do so.
Although data participants should have the right to withdraw their consent for data sharing, the practical difficulties in implementing this should be made clear.
There is no dispute that the right to withdraw consent to data sharing must be respected. In legal terms, the need for a consent is normally coupled with a corresponding right to withdraw that consent, and this is acknowledged, for example, in the GDPR (article 7.3).50 As long as the stored data are still pseudonymised (ie, a participant’s data can be identified), a participant’s request that their data be removed from the data set can be honoured. This might involve providing new versions of data sets to repositories, and be supported by including clauses about the management of withdrawn consents in data use agreements.53 As pointed out in the EU clinical trial regulation 536/2014, however, the withdrawal of informed consent should ‘not affect the results of activities already carried out, such as the storage and use of data obtained on the basis of informed consent before withdrawal’ (paragraph 76 of the preamble).51
The practical difficulties, and associated costs, in modifying data already delivered to a separate repository should not be underestimated, and it may therefore be difficult to offer the withdrawal option once data have been deposited. There are even more difficulties in withdrawing data after they have been shared with a secondary user—in fact this may be impossible in practical terms. The key point is that any limitations to withdrawing consent for data sharing should be made clear in any explanatory material in the patient information sheets.
Data preparation: protection of trial participants
P3: IPD made available for sharing should be prepared for that purpose, with de-identification of data sets to minimise the risk of reidentification. The de-identification steps that are applied should be recorded.
Shared IPD from clinical trials used for further scientific research should always be de-identified and either pseudonymised or anonymised (see glossary in online supplementary appendix 2). All three are important concepts, although only the last two are used within EU law. Any consideration of data preparation requires a shared understanding of these terms, so they are discussed below.
De-identification is not defined under the GDPR but is defined in the USA, for example in the Health insurance portability and accountability act (HIPAA) regulations.54 It means removing or recoding identifiers, removing or redacting free text verbatim terms, and often removing explicit references to dates. Participants’ identification code numbers are de-identified by replacing the original code number with a new random code number. It is used in this document to indicate that identifiers have been removed from a data record but does not necessarily mean that the data record meets the requirements of being pseudonymised or anonymised according to GPDR.
Pseudonymisation means processing personal data in such a way that the data can no longer be attributed to a specific data subject without the use of additional information, (eg, a data set linking trial identifiers to identified or identifiable persons) provided that such additional information is kept separately and under controlled access, to prevent the data being identifiable in isolation. Although theoretically such information could be used to match against a clinical trial data set and identify individuals, this would be very difficult in practice and could only occur if there was a major breach of security.
Anonymisation is a technique applied to personal data to make it, in practice, unidentifiable. Full (complete, or irreversible) anonymisation involves de-identification and the destruction of any link to an identified or identifiable person via a pseudonym. Effective anonymisation can be applied to a specific data set, by de-identification and removal of the link to a pseudonym, coupled with the use of new identifiers for individuals. There is no link maintained between these new internal identifiers and any others that might exist, for example in another pseudonymised data set (eg, pseudonymised data set of the sponsor).
Thus, if a de-identified data set is pseudonymised, the participants in it can be identified only by those who possess the relevant ‘additional information’. If a de-identified data set is fully anonymised, the participants cannot be identified by anyone (leaving aside the theoretical possibility of matching against the original clinical data). If a de-identified data set is effectively anonymised, there remains only the very small possibility of matching the data against a corresponding but pseudonymised set, if it is accessible (it should not be), but the matching cannot be guaranteed, especially if the participants share many of the same data values.
Before data can be shared, they should be de-identified, removing possible identifiers to minimise the risk of reidentification.
Adequate de-identification is one of the key determinants of successful protection of study participants from reidentification. The level of de-identification required for both pseudonymised and anonymised data is the same. In all cases it should provide a high level of assurance that the data content, in and of itself, cannot be used to identify the individuals within the data set. Other policies and procedures (eg, the use of a data use agreement) also provide protection against reidentification, but de-identification is a necessary prerequisite and should be applied to all data made available for secondary use.
Shared data should remain pseudonymous unless that is not allowed by the relevant legislation. Additional information that may allow reidentification should be stored securely and not shared.
Sharing of pseudonymous data is recommended and should be the normal expectation. Clinical trial data are pseudonymous when collected, or can be easily turned into pseudonymous data within the research unit, by processing of the data set and splitting off the identifying data points. It would be rare for trial data to become fully anonymised, or at least not until many years have elapsed after data collection. There are legal obligations on sponsors to maintain the pseudonymised data set, as collected, for many years, the exact time depending on national regulations. In addition, the original investigators, or their institution, may want to use the pseudonymising key in case they wish to return to the same participants to carry out further investigations (assuming they have the ethical approval and/or explicit consent to do so).
The principal options for sharing data are therefore (1) to share the pseudonymous data set, but not the pseudonymising code; or (2) effectively anonymise the data set before it is shared, by replacing the identifiers used in the trial with another independent set and not retaining any linkage information between the two.
The advantage of sharing pseudonymised data is that, if the secondary user discovers good reasons for clarifying, expanding or matching some of the data, or even for further investigations with some of the source population, they can contact the holders of the pseudonymous data and discuss if and how this might be achieved, because the individual participants are still (indirectly) identifiable. This does not mean that identifiable or identifying information would be transferred to a secondary user, unless there was explicit consent from the participant for this to happen (although this seems unlikely to be given). It only means that if a case can be made for identifying the individuals in the data set, it is at least possible to discuss the possibilities of doing this, including possibly returning to the individuals concerned to request additional consent.
Standard procedures and techniques for de-identification should be applied, whenever they exist, and fully documented to ensure transparency and reproducibility.
De-identification should be consistent with current standards, guidelines and policies provided by official bodies and scientific organisations.55–62 Techniques and guidelines for de-identification of health data exist and are becoming more common in research (eg, ref 63). The record of de-identification should be stored, most usefully alongside the de-identified data set as another piece of metadata. To make it easier to review the de-identification that has occurred, we need a standardised, and ideally machine readable, way of describing those de-identification actions.
An assessment of the residual risks for reidentification of participants in de-identified data sets should be performed.
Under the GPDR, at least in Europe, there is obligation on the data controller to carry out a data protection impact assessment (DPIA), to ‘evaluate…the origin, nature, particularity and severity’ of the ‘risk to the rights and freedoms of natural persons’ before processing personal data. The impact assessment ‘should include the measures, safeguards and mechanisms envisaged for mitigating’ the identified risks. This implies that the initial de-identification of data, for instance prior to its deposition in a repository, should be accompanied by such an impact assessment, ideally included within the record of de-identification described in recommendation 17.
In addition, at least in a managed access environment, assessments of reidentification risk should be made when data are requested for secondary use, because a full risk assessment will be sensitive to the particular context of the planned usage, in particular any data use agreement. If the data have already been adequately de-identified, such a risk assessment may be relatively light, and in some cases, may be delegated to the repository managers.
Practical guidance is available on managing de-identification and assessing the associated risks. For example appendix B of the Institute of Medicine’s paper on data sharing,14 ‘Concepts and Methods for De-identifying Clinical Trial Data’, provides a useful overview of both the assessment of risks and strategies to mitigate them, focused on but not restricted to the US context. In Europe, the article 29 data protection working party has produced a detailed guide about the DPIA and how it should be applied.64 But it should be noted that, at this point, it is unclear how different national jurisdictions may interpret the requirements for impact assessment in the specific context of the sharing of clinical research data. The legal responsibilities of the trial sponsor, as the data controller, and if and how they might be delegated to others, remain to be clarified.
Reidentification of data subjects should always be forbidden.
Attempted reidentification of data subjects should be explicitly prohibited in any formal data use agreement. Even when a binding agreement does not exist, attempting reidentification is likely to be illegal, and in any case should be subject to sanction. The sanctions that might be applied could be organisational (eg, for serious misconduct) and financial (eg, loss of access to further funding) as well as legal (eg, for breach of contract).
In cases where no explicit consent for data sharing was obtained from the trial participants, data sharing may still be possible if the data are prepared, and data requests processed, in ways that maintain legal compliance.
Data that do not carry an explicit consent to data sharing (as from many past and current trials) could still be shared in circumstances where national or other regulations allow for exceptions to the normal restrictions on data sharing, for instance where obtaining consent is seen as too impractical for researchers or too burdensome for participants, and the risks are assessed as low. In such circumstances, it is anticipated that the proposed sharing request and data use may need the involvement of ethical committees or other review boards, dependent on national systems. In addition, the data may be required to undergo an increased level of de-identification, and the data use agreement may impose greater restrictions on data access.
Effective anonymisation may also be an option, although there has to be a mechanism to agree that anonymisation has been truly achieved. If that is the case the data protection regulations no longer apply. Anonymising data will itself usually be seen as data processing, and thus covered by data protection regulations. The anonymisation would therefore have to be done by someone who had been authorised to process the data.
The difficulty is that many of the issues surrounding the secondary use of data without explicit consent have yet to be clarified, and will need (in Europe) the further interpretation by national authorities of the requirements represented by the GDPR, in the specific context of clinical research data. The emphasis in future trials should be on avoiding this issue altogether, by a rapid and widespread introduction of explicit consent procedures for data sharing.
Services to support de-identification of data sets, which could range from simple guidance, through consultancy, and on to performing and documenting the de-identification process, should be established.
To ensure good practice in this area, it would be useful to identify existing centres of expertise and/or develop central services that could provide robust de-identification practices, documentation and/or review. Such services could make use of the existing guidelines and good practices, as for example those from the Council of Canadian Academies,61 and develop them further in the particular context of clinical trial data. In time, such good practices could be disseminated to research units so that they become able to carry out their own de-identification measures.
Data preparation: data standards
P4: To promote interoperability and retain meaning within interpretation and analysis, shared data should, as far as possible, be structured, described and formatted using widely recognised data and metadata standards.
A greater use of data standards is critical to the success of data sharing. Without such standards, any shared data are harder to interpret with confidence and much more time-consuming, and thus costly, to aggregate. Standards can apply to data item definitions and codes, to controlled vocabularies used for categories, and even to the way data are structured and exchanged. The file formats used for storing and transferring data should also be standardised to make data processing easier.
It is accepted that the nature of clinical research, where novel interventions may be under test, means that it may sometimes be necessary to create new definitions and codes for some of the data items used in a trial. The aim, however, should be to make use of widely recognised data standards wherever possible (such as those from CDISC). Where new definitions are required, to support new science, they can and should be derived by extending existing standard schemes. The widespread use of data standards has a critical role in reducing the costs and maximising the utility of data sharing.
Data and coding standards should be built into any trial’s data design prospectively, from the beginning of the trial.
It is very difficult to try and apply standards and data definitions after a trial database has been designed and the data collected, or to try and change data structures unless a trial has been designed from the beginning with those data structures in mind (eg, it is much easier to map data to Clinical Data Interchange Standards Consortium - Study Data Tabulation Model (CDISC SDTM), the tabular data format used by the Food and Drug Administration (US) (FDA), if it has been collected using Clinical Data Interchange Standards Consortium - Clinical Data Acquisition Standards Harmonisation (CDISC CDASH) data items). Legacy data conversion can be done when there is value in combining data from prior trials, but it is resource-intensive and may compromise data integrity. The time and costs required for retrospective ‘standardisation’ would put such an exercise beyond the resources of many non-commercial units. Instead, it is important that standards are designed from the start, with decisions made about the coding and other systems to be used made as part of the trial design process.
Among the various data standards available, those from CDISC should be considered as offering the best starting point currently available for defining and coding data and metadata in a consistent way.
In a steadily evolving standards environment, there is clearly a risk attached to recommending any specific standards. Nevertheless, the work CDISC has done in developing standards in clinical data items and data structure for nearly 20 years has resulted in a suite of useful and harmonised data standards of particular relevance to clinical trial data.65 We would encourage researchers to examine one or more of these standards, which have been widely adopted around the globe, as a vehicle for introducing more standardisation into their trial data. Of course, using other recommendations and standards—for example, core outcome sets as collected by COMET,66 Medical Dictionary for Regulatory Affairs coding for adverse events67 and the eTRIKS standards for translational research68—can also increase interoperability between data and complement the CDISC standards.
It will also be important to develop standards further so that they can apply to a greater proportion of data from clinical practice, including working towards a maturation of healthcare data standards, such that they can be used synergistically with research standards.
Non-commercial clinical research infrastructures should actively support the prospective use of data standards, for instance by taking advantage of existing training, materials and supporting services, and expanding these as needed.
The use of data standards in non-commercial research has been relatively limited up to now, and consequently there is a need to increase awareness of the different standards available and their uses, and develop tools and services that can help researchers apply them in practice. Infrastructure organisations, such as ECRIN and the various national networks, working with the standard development organisations, can play a key role in this. Support might range from awareness raising workshops and developing informational materials through to curating libraries of data collection instruments. For CDISC standards there is SHARE (Shared Health and Research Electronic Library), a tool providing access to curated machine-readable versions of CDISC standards and terminology.69
Non-commercial clinical research infrastructures should actively participate in the standards development process to further extend the standards as needed.
There is a need for more non-commercial research organisations and infrastructures to become involved in data standard development. In the past standards development has often been driven by requirements for submission to regulatory authorities, although, more recently, the process has broadened to encompass standards that apply to public health and disease outbreaks, nutrition research and observational studies.
It will be important to continue these developments to ensure that standards are equally useful, and equally applicable, to both the commercial and non-commercial research sectors. We recognise that increasing the engagement of non-commercial research facilities with data standards will necessarily be a gradual and long-term process, but the potential scientific benefits are too great for that engagement not to occur. Key to that process will be academic recognition and reward for input into standard development.
Clinical trial data sets should always be associated with metadata that describes the characteristics of each data item (eg, type, code, name, possibly an ontology reference), as well as the schedule and design of the trial.
As a minimum, a basic data dictionary and study schedule should be provided, for instance as spreadsheets, or as an (CDISC) operational data model (ODM) XML file. Ideally, however, the metadata should include the meaning of the individual data items (eg, to clarify different types of blood pressure measurement, or the meaning of ‘clinically significant’) either by providing brief descriptions or by referencing a published ontology. The CDISC Define.XML metadata system provides one mechanism to remove ambiguity in this way. The more uniform data set metadata becomes, the more feasible it will be to build tools that can search, compare and aggregate data sets automatically, potentially reducing the costs of data reuse.
Data sets should be made available for sharing in one or more standardised file formats that can be read by a wide variety of different systems.
Proprietary and statistical software formats should be avoided. Using relatively simple and generally interchangeable file formats (sometimes referred to as transport standards), which can be accessed using a variety of file manipulation tools, is an important aspect of making shared data as accessible as possible to a wide range of potential users.
Any formats should, however, allow for the explicit preservation of structure within the data, including parent–child relationships. For that reason, structured text, based on XML schemas, is a particularly useful and generally applicable format. ODM XML has the advantage of supporting an audit trail to ensure data traceability and provenance.
Rights, types and management of access
P5: Access to IPD and trial documents should be as open as possible and as closed as necessary to protect participant privacy and reduce the risk of data misuse.
A range of access types to shared data and documents is expected and encouraged, including different forms of controlled access.
The guiding principle we encourage is that IPD and associated documents should become as openly accessible as possible. Although we believe most trial documents should be openly accessible without restrictions, we acknowledge that IPD may pose concerns for the data controllers (the sponsors)—over protecting participant privacy—and the data generators (the investigators)—for instance over possible misinterpretation of the data. Given the current lack of established standards surrounding IPD sharing, we believe a range of access models to data sets will be inevitable. We would recommend, however, that for IPD the secondary user should as a minimum identify him or herself, and agree to some basic conditions of data use (see recommendation 29).
Depending on several factors (eg, the nature of the consent obtained, risk of reidentification, concerns about stigmatisation, misuse of information, incorrect analysis and so on), access models may range from publicly accessible web-based systems, with the possibility of downloading data sets, through various types of request/review mechanisms that may or may not allow data download. A granularity of access may also be applied to different parts of the same data sets, as some piece of information may be more sensitive or difficult to handle than others.
We acknowledge that the issue of who is responsible for choosing one access model over another is not yet resolved. Data generators will usually be most familiar with the potential value of the data, as well as the risks associated with its misuse, so should have a role in the definition of access schemes. Data repositories may also have a role in this process, if some or all aspects of access control have been delegated to them by the data controller. The final goal should remain, however, the maximisation of the value of data. It would therefore be useful to establish mechanisms to monitor data access regimes, and where necessary to identify and help modify any overprotective schemes.
Access to IPD should always be accompanied by a statement of compliance with basic rules designed to promote a fair sharing of data.
We believe that all secondary data users should acknowledge and agree to some basic rules of data use. For instance, they should identify themselves (including validating their email address using a call-back and confirmation process), not attempt to reidentify participants, make the results of any secondary analyses public and cite the data source correctly in any published work. The definition of international standard practice for data sharing would usefully clarify these basic rules, and help to alleviate the fears of researchers about possible problems. At its simplest compliance with the basic rules of reuse could be signalled by completing a web-based form. More detailed attestation or formal agreement is likely to be needed in some situations, for example if the original consent to secondary use mentions possible restrictions, data sensitivity is high, or the data generators are concerned over misinterpretation.
We acknowledge that some data repositories currently host de-identified clinical trial data sets that are available for immediate perusal or download without any type of restriction or registration.70 71 Although this is clearly possible, we reiterate that the secondary user should normally be asked to comply with some core principles, as an important aspect of maintaining the transparency of the data sharing system and making data sharing more acceptable to all stakeholders.
Boards overseeing the data sharing process may be established, ideally at the level of data repository. These boards may provide advice on ethical and legal issues that may arise in data sharing and, for controlled access, may be responsible for the management of data access requests.
The presence of a board that oversees the overall data sharing process and, if applicable, evaluates data access requests has been widely advocated. The role and responsibilities of such boards may vary. As an initial step, we envisage the creation of boards of experts (‘access advisory committees’ or some equivalent term) who can provide advice and support to data generators and repositories. Ideally, these boards would be established by repositories or groups of repositories.
In the same way that data generators are encouraged to use suitable repositories for storage, and for the same reasons of providing continuity of data management in the longer term, we encourage the delegation of access management to the repositories and their boards. When a controlled access model applies and a formal evaluation of the data request application exists, we encourage a process where the assessment of the scientific merit, potential impact and appropriateness of the proposed secondary analyses is performed by independent data access boards. These boards could also assess and ensure that the data generators were fully cited and recognised, although this would only work if mechanisms to track citations and highlight when recognition was not given were in place.
Irrespective of the tasks delegated to these boards, transparency in their mandate, procedures, composition and expertise is essential.
Whatever the exact mandate of any particular board, it will be important that its work is transparent and that its membership is known. It is important that any board includes a wide range of expertise, including representatives of citizens and patient groups. Any possible conflicts of interests (including non-financial ones) should be declared and managed. The evaluating criteria and process should be public, as well as aggregated metrics about the reasons for accepting and rejecting particular requests. This will ensure the transparency of the decision process and be of aid to future applicants.
P6: In the context of managed access, any citizen or group that has both a reasonable scientific question and the expertise to answer that question should be able to request access to IPD and trial documents.
The right to request access to data should not be limited to specific professions or roles.
As a general principle, access to data should not be limited to a specific type of requester or professional profile. In cases where the access model includes a formal evaluation of a data access application, the scientific question to be addressed, and the ability of the requesters to answer that question, is more relevant to the assessment of data requests than the requesters’ current job roles. Data could be sought, for example, by students and science journalists as well as by active researchers or reviewers. The requesters or their team would, however, normally need to demonstrate the ability to draw scientifically literate conclusions from the data.
If access is formally managed, the data requester may need to provide a research protocol and analysis plan, including information on data management, data storage and plans for publication of the results of the reanalysis. The requester should also provide information on his/her (or team) expertise, possibly making use of persistent digital identifier systems (eg, open researcher and contributor ID (ORCID)).
Consideration of access requests should not, in principle, be influenced by whether the proposed secondary reuse is associated with a potential commercial benefit, directly or indirectly, in the short or the long term. There is, in any case, often difficulty in clearly differentiating ‘pure’ from ‘applied’, or ‘commercial’ from ‘non-commercial’ research.
Collaboration between data providers and secondary data users could be an added value in data sharing. However, it should not be a prerequisite for data sharing.
Several benefits can arise from the involvement of the data generators in the reuse of data. The original investigators can share key insights with the secondary users about the study, its data and analysis, reducing the possibility of misinterpretation of data. This kind of cooperation may therefore substantially enhance the quality of secondary data usage and make it more efficient.
In the model of data sharing envisaged in this document, there is, however, no necessity to involve the data generators (as was often the case in the past, when data were shared within research collaborations), and whether such involvement is planned should not influence, in a controlled access environment, the data access decisions. If there is active participation by the original data providers, then coauthorship in the publication resulting from the reuse will normally be appropriate, following the established rules on authorship.40
Even if not directly involved in the secondary use, it is reasonable that data generators (assuming that they have not made the data access completely open) should have the option of being informed about who is accessing data, or requesting such access, and when. This would be possible if secondary users are always asked to identify themselves (see recommendation 29) and could be part of a formal agreement between data generators and repositories (see recommendation 42).
The results and methodology of further analysis of data and documents should themselves be publicly available and deposited in an appropriate repository, whether or not they are associated with published papers.
Data users should agree to make the methods and results of their secondary analyses publicly available not only through scientific publications (that may or may not be prepared and, if prepared, that may or may not be accepted for publication) but also by depositing them in a repository and making them discoverable. This will be important to provide further examples of effective data sharing and allow any conclusions from secondary use to be examined by others.
P7: The processing of data access requests should be explicit, reproducible and transparent but, so far as possible, should minimise the additional bureaucratic burden on all concerned.
Within a formally controlled data access system, that is, one requiring explicit request and evaluation of that request, the process through which data can be accessed should be clear, reproducible and transparent. Inconsistent decisions should be avoided and criteria should be explicit.
To simplify the request process, repositories should be encouraged to make the interface presented to secondary users as consistent as possible.
Processes, information requirements and proformas should be the same or very similar between different repositories, to make life simpler for all concerned but especially the secondary data users. It may even be possible to develop a common ‘access request pipeline’, especially for smaller repositories, so that associated costs could be shared, even if each repository retains the rights to individually approve or reject requests.
Taking this one stage further, it should also be possible to share boards across repositories. The existing CSDR scheme provides a similar approach, with data generated and stored by different commercial companies, but with the Wellcome Trust orchestrating the process and supporting a common Independent Review Panel.23 There are questions about how such a scheme could be funded in the non-commercial domain, and how the membership of a common review board could be made acceptable to many different users. Despite these issues, however, this approach could offer considerable simplification for secondary users, and reduce the bureaucratic burden on repositories.
The implementation of a standard terms of use agreement, a ‘data use agreement‘, specifying the conditions for data access and reuse, is encouraged.
Such an agreement should not constitute an obstacle to data sharing—instead it should facilitate it by ensuring that the rights, roles and responsibilities of all parties are defined.
Templates for data use agreements (along with an explanation for the information requested) could be developed, made public and shared by several repositories to simplify the access request process.
An appropriate data use agreement should include at least the following aspects:
Partners and bodies involved.
Clearly identifying the parties and their role and responsibilities.
Definitions.
Where there is any real or potential ambiguity, terms should be defined.
The purpose of the request and possible restrictions.
A description of the intended, agreed use and any limitations to that use (eg, restricted to research in a particular disease area). This section should also include definitions of inappropriate use of data and any restrictions on how the data can be used (eg, distribution of data to third parties, attempt to reidentification).
Agreement to acknowledge and give credit to the original data generators.
Public dissemination of the results of the reanalyses.
An agreement to provide public deposition of results, often but not necessarily in the same repository as the source data.
Consent issues.
How consent for IPD sharing has been managed, for example, a description of the consent being used to justify the data sharing (or in the absence of explicit consent a description of the regulations under which sharing is taking place and how they have been met).
Terms and conditions of control over the data within the requesting organisation.
How the data will be managed and stored in the organisation of the requester(s), assuming a data download, and the measures to be taken to ensure appropriate access and security.
Terms and termination of the agreement.
Defining the period during which the agreement is effective. Specifying the conditions under which the agreement can be terminated before the contractual duties have been fulfilled (eg, breach of data sharing code of conduct and so on). How the data will be managed once the agreement is terminated (eg, will data be returned to the provider or destroyed?).
In order to allow data sharing as open as possible, unnecessary restrictions due to intellectual property issues, patents and licences should be avoided. Data and objects should be deposited in repositories under licences that maximally support data sharing, for example, with Creative Commons, that allow others, in legal terms, to both use "works" and to make "derivative works" from them.
Tools should be developed to support the implementation of common metrics across different data sharing platforms and repositories, publishing these under a common portal.
Examples include the numbers and types of request and approval data, together with reasons for not providing access, and summary data (including links) on the published papers resulting from reuse of data and documents. This is an important aspect of maintaining transparency across the entire data sharing process.
Mechanisms to collect and display user feedback, about the process of accessing data or data sharing in general, should be developed and implemented by repositories themselves or by third parties.
Such feedback could be a useful complement to the data described in recommendation 38, helping to improve transparency and increase user involvement, as well as providing direct feedback to repositories. Implementation could be by individual repositories or by an external service, or by some combination of the two.
Data management and repositories
P8: Besides the IPD data sets, other clinical trial data objects should be made available for sharing (eg, protocols, clinical study reports, statistical analysis plans, blank consent forms) to allow a full understanding of any data set.
In any discussion about data sharing the emphasis is naturally on the data sets themselves. But to fully understand that data requires the context, purpose and timing of the data collection to be clear, as well as the processing and analysis that were originally carried out on that data. That in turn demands that protocols, analysis plans, study reports, case report forms (CRFs) and so on also be available for sharing, and need to be managed as available ‘data objects’—preferably within designated repositories (as in the following principle 9). If not, there is a danger that the data, considered in isolation, could be misinterpreted. For both data generators and secondary users, therefore, it is important that the material that needs to be stored and managed, and potentially shared, includes all relevant documents as well as data sets.
P9: Data and trial documents made available for sharing should be transferred to a suitable data repository to help ensure that the data objects are properly prepared, are available in the longer term, are stored securely and are subject to rigorous governance.
There is a risk that ‘making data available for sharing’ could be interpreted as the original research team simply agreeing to consider data requests on an ad hoc basis. We feel that there are several problems associated with this, however, and that the alternative—of data being transferred to a designated data repository—is a much better option. The reasons for this include:
The original research team (or collaboration) will change its composition, or may even cease to exist, and it may then become difficult or impossible for data to be managed and requests to be properly considered.
The transfer of data to a third-party repository makes it more likely that preparation of the data for sharing (eg, de-identification, provision of metadata) will occur, and help ensure that the data and related documents are properly described.
Planning for transfer to a repository helps to explicitly identify data preparation and sharing costs at an early stage of the trial.
It helps to make the data and trial documents more easily discoverable.
It can relieve the original research team/sponsor of the need to review requests and even (depending on the arrangements made with the repository) of the need to make the decisions about agreeing to such requests.
A ‘designated data repository’ in this context may be one dedicated to clinical research data and documents on a global or regional level, a general scientific repository, or one specialised in storing data objects related to a specific disease area. It may be a repository established by the researchers’ own institution for ‘their’ research. We make no recommendations about the optimum scope of a repository—only about the processes it employs.
Repositories for clinical data and data objects should be compliant with defined quality criteria.
The services any repository provides should conform to specified quality standards, to give its users confidence that their data and documents will be stored securely and in accordance with the specific data transfer agreements they have agreed. Some generic standards and criteria for trustworthy digital repositories have been developed and are being applied (eg, Data Seal of Approval,72 International Council for Science World Data Systems,73 DIN 3164474) and several instruments for certification of repositories have been implemented.72 73 75 76
The necessity for collaboration and harmonisation of these different activities has been acknowledged,77 and proposals for a unified core set of requirements for trustworthy data repositories have recently been made (International Council for Science/World Data System (ICCSU/WDS), data seal of approval (DAS)78). The available standards, requirements and certification instruments for trusted data repositories need to be examined and their applicability to clinical research data objects needs to be checked. If necessary, extensions or adaptions should be provided.
There will also be a need to develop or adapt sustainable systems to assess repositories for clinical data and data objects against these standards. This is all work still to be undertaken but, given the likely variety of repositories that will be available to researchers, we see it as a necessary part of any acceptable data sharing environment. Research infrastructure organisations can play a key role in developing and disseminating both the standards and the assessment systems.
Information about the different repositories that hold clinical research objects should be made available to data generators so that they can make an informed choice, so far as local policies allow.
This information should include costs as well as the features and access options available, and any assessment against the quality standards described above. The purpose is simply to assist the data generators in their decision on where to store data objects, as well as to encourage some healthy competition between repositories. We envisage a central service giving information and contact details on the repositories available, similar to the data provided now by re3data for general repositories (we believe the current re3dataset would need substantial modification to support the needs of clinical researchers selecting repositories). Ideally the repositories themselves would find it beneficial to keep their records within such a system as up to date as possible.
The transfer of any data objects to repositories (including those within the same institution) should be subject to a formal agreement that set out the roles, rights and responsibilities of the data generators and the repository managers.
We would expect a data transfer agreement to apply to the transfer of data and documents to a repository. In other words, the transfer should always be a formal arrangement, with the responsibilities of each party clearly set out, rather than an informal upload. Aspects that are particularly important include the agreed access regime for the data, the mechanism by which any future data sharing decisions will be made and the assignment of the data controller role.
Mechanisms for implementing an ‘analysis environment’, allowing in situ analysis of data sets but preventing downloads, should be further evaluated. Such an analysis environment should allow different data sets from different host repositories to be combined on a temporary basis
This would be a specialist repository facility analogous in many ways to the ‘glove boxes’ or analysis environments now made available for examining some pharmaceutical research data. The process would include:
Gaining permissions for the temporary ‘loan’ of data sets from different repositories into the analysis environment.
Setting up a temporary IT system (virtual machine or container) with the necessary analysis tools included.
Importing the data sets as agreed.
Carrying out and recording the analysis.
Gathering the results.
Destroying the temporary IT system and the data it contains, usually straightaway but in any case, according to prior agreement.
The advantages:
It gives the repository/data generator greater control over control of access and may therefore encourage wider and/or earlier data sharing.
It allows the aggregation of data from widely different sources, more quickly than could be done by multiple applications to download files.
The disadvantages:
It demands a more complex and expensive technical infrastructure, including a much greater degree of human input for each data aggregation, than a system based on simple downloads.
It requires trust between the repository/data generator and the organisation providing the facility, for example about the security and access controls in place.
There are also several non-trivial challenges that need to be overcome if this type of facility is to work at scale:
Stable application programming interface (APIs) need to be developed that allow data retrieval and access across multiple repositories.
Data standards need to be applied that allow interoperability of the retrieved data.
Cloud environments need to be constructed with appropriate security, audits and account management.
Transinstitutional (some of which may also be transnational) cost sharing and accounting models will be required.
These are issues being addressed in other scientific domains, however, and they should not be insurmountable within clinical research.
Discoverability and metadata
P10: Any data set or document made available for sharing should be associated with concise, publicly available and consistently structured discovery metadata, describing not just the data object itself but also how it can be accessed. This is to maximise its discoverability by both humans and machines.
We believe that there will be many different repositories used for clinical research data objects, complementing the existing systems used to index peer-reviewed papers and the registries that include details of the trials themselves. We therefore need mechanisms to support discoverability across this mosaic of resources. Reviewers and researchers need to be able to identify the data and documents related to a trial, and discover how they can access them, and the restrictions on use, in an efficient and consistent way. A metadata description of each individual data object is key to that requirement, as it provides a means by which software agents can interrogate different repositories and aggregate their ‘lists of contents’, to form a single source of information.
A metadata schema suitable for describing all repository data objects linked to clinical trials needs to be developed and implemented, agreed by major stakeholders and repository managers and widely disseminated.
Such a schema should include clear identification of the source trial (or trials) and of the access arrangements that apply, as well as a description of the data object itself. Within the CORBEL project, proposals have been made based on the widely used DataCite standard,79 but any such schema requires further discussion by repository managers and others, with the goal of agreeing a common standard.
Repositories with clinical research data objects should use this generic schema for those objects, or a schema that can be easily mapped to it, so that the metadata describing the contents of different repositories can be aggregated.
This is an ambitious goal because of the global scale required (to be really useful all sources of data objects need to be included), but it is difficult to see how any discoverability mechanism can be made sustainable in the longer term without a generic schema being used. The alternative would require a range of aggregation/reconciliation techniques for different types of metadata, and/or need to use ‘data mining’ techniques to link records. This may be an option for legacy trials, but is of limited value in the longer term because it is likely to be too difficult, error-prone and costly other than in a pilot or research project. We therefore need the widespread use of the schema described in recommendation 44, to allow automatic and reliable aggregation of metadata.
The generic metadata scheme will need to include a common identifier scheme for clinical research data objects. The DOI is recommended as the best candidate for such an identifier. Mechanisms should be developed to make it easy to assign unique identifiers to all data sets and documents that are made available for data sharing.
Any metadata schema needs at its core a way of assigning globally unique persistent identifiers to the objects being described. The DOI seems to be the most appropriate identifier to use for this, not least because so many existing data objects and published papers make use of the same mechanism. Allocating DOIs will have to be done as cheaply as possible, and various mechanisms, perhaps using the existing abilities of some universities to assign DOIs, or involving infrastructure organisations as the source of DOIs, need to be explored to identify the most effective approach. A related issue that needs to be tackled, although it is outside the scope of the CORBEL project, is the allocation of unique persistent identifiers for trials, although various ‘workarounds’, for example, the use of registry IDs, are available at the moment.
Tools should be developed to help data generators to complete the metadata fields of the generic scheme described above as efficiently as possible.
One could envisage a web-based system that provided the necessary fields and prompts and that could be made available to data generators. It is important that wherever possible it is the data generators that create the metadata, as only they have the full knowledge of the material required (although they might not provide the metadata until the data objects are about to be transferred to a repository). The advantage of web-based data collection is that it could also aggregate the data for different repositories at the same time, because the data would be stored in the same ‘back end’ database system. This would then make it much easier to make the data available through a single portal.
Tools should be developed to enable the regular harvesting of metadata data from repositories, importing that metadata into a collection of ‘metadata repositories’ for clinical research data objects.
As stated above, this is a key component of aggregating metadata into useful collections. Data that are not generated centrally will need to be imported regularly, for example by using APIs to ‘harvest’ the metadata at regular intervals (eg, daily). The more diverse the metadata the more difficult the task, and initially a range of such tools might be required. Over time, if the metadata becomes more consistent as described above, the software systems can themselves become simpler and cheaper to maintain.
Metadata repositories should be developed, sustained and connected to enable common web-based access portals to the underlying metadata, providing a single point of entry for users as well as associated search facilities.
The broader the scope of a metadata repository the more useful it is to its users. The concept here is of a global metadata repository (MDR) portal, that is, website, connected to a range of individual metadata stores maintained by different stakeholders. If the metadata used has a consistent schema across the various systems, then the whole aggregation of data becomes searchable as a single resource.
Mechanisms to sustain metadata repositories and the portal/search systems that connect to them in the long term should be developed, based on the recognition of the importance of such services for data sharing.
The discoverability mechanisms described in this section are of little use unless they can be sustained permanently. Pilot metadata repositories should be established (and existing initiatives, eg, OpenTrials,80 supported) to allow clearer identification of costs and the issues with running such a service. The research community and governments then need to agree funding mechanisms and infrastructure (eg, within the developing EOSC) that can support discoverability in the longer term.
Discussion
The debates around sharing and reuse of IPD from clinical research have expanded rapidly in recent years, reflecting the fact that there is now wide agreement that it will benefit research and thus, eventually, healthcare. However, many questions concerning principles and practice remain to be resolved. For instance, how to best promote and support data sharing and reuse among researchers, how to adequately inform trial participants and protect their rights, and how, where and in what format data should be stored, found and accessed.
This document has discussed a number of these questions, using an approach based on the ‘life-cycle’ of data sharing. It articulates 10 principles, developed by the multistakeholder group of international experts after a formal consensus exercise, that represent an overarching framework for IPD sharing and reuse. The framework has been further developed into 50 more detailed recommendations to provide what we believe to be clear practical guidance on how best to make data sharing work.
Methodology
To tackle an issue as complex and multifaceted as sharing IPD from clinical trials, we first established an international group of experts covering a broad spectrum of expertise and experiences from different areas (trial methodology and registration, research transparency and ethics, meta-analyses, scientific publisher, regulatory bodies, patient organisations, data protection and IT experts, standardisation bodies, and IT service providers).
Second, we applied a standard methodology for consensus elaboration, that is, a nominal group process with the support of an independent facilitator. The group attended three face-to-face meetings over 1 year with excellent participation, extensive discussion time and a structured decision-making process. The nominal group process gave all members of the task force the opportunity to identify issues and then for the whole group to debate and vote on them.
One major issue was evident from the beginning of this consensus exercise. The terminology around data sharing is confusing and, often, the same term is used by different stakeholders or in different contexts (or countries) to point to different concepts. For instance, different understanding of terms such as ‘anonymised’, ‘pseudonymised’, ‘de-identified’ or ‘metadata’ impaired discussion at times. For this reason, the group developed and agreed on a glossary (online supplementary appendix 2) to be used in the context of the discussion, which hopefully can be useful as a general reference.
Contentious issues
Consensus did not always mean unanimity. The group reached a common view on general principles relatively easily, while, as expected, some of the detailed recommendations raised more discussion. In only a few cases, however, were clearly divergent positions held by more than a small number of task force members.
One was the issue of whether the consent for data sharing needs to be distinguished from the consent to participate to the trial. It was acknowledged that a separate consent is often required by law, particularly in Europe, but a conception of data sharing as an integral part of the clinical trial process prompted a substantial minority of the group members to propose a single consent mechanism: to participate in the trial and to share pseudonymous individual data. The reasoning behind this position was that, ultimately, data sharing and reuse are intended to help improve the health of all, and the utility of data sharing is increased if it encompasses all trial participants. At the heart of the debate was the different emphasis people put on the autonomy, privacy and safety of the individual, versus the potential gains to society from increasing the ease and efficacy of data sharing. The majority in the task force felt, however, that distinct consents were necessary, and in any case a single consent process would be hard to implement within the current legislative frameworks, at least for pseudonymised data (see recommendations 12 and 14). Nevertheless, it was clear that this issue raised considerable and passionate debate, and that it deserves more detailed research and discussion involving experts in medical ethics and law, researchers, trial participants and citizens.
A related issue is the question of whether, in general, the data that are shared should be pseudonymous or anonymous. As explained in recommendation 16, the preference of the task force was for the former, although anonymisation of data will be necessary where no explicit consent for data sharing has been obtained (see recommendation 20). An argument was made that the sharing of anonymised data should be the norm as it is likely it will make data sharing more practicable and quicker to establish. It may be that the early years of data sharing will require much greater use of anonymised data, until explicit consent for reuse becomes more widespread. The question is whether this could impact the scientific utility of the data, largely in terms of the potential for follow-up work (the degree of de-identification should be the same for both anonymised and pseudonymised data). This will require further empirical investigation.
Our findings in context
In recent years, several other organisations and projects have developed principles and recommendations for IPD sharing, as summarised in table 1.
Main initiatives aimed at developing principles and recommendations for IPD sharing
The output of our consensus exercise therefore fits into a context of earlier initiatives embedded in specific national or geographical contexts, or dedicated to specific stakeholders. We believe that by providing a pan-European perspective on the issue of IPD sharing and reuse, and by looking at all aspects of the data sharing ‘life cycle’, the current document is a useful addition to the previous work in this area, complementing the reports centred on the USA, the UK or the Nordic countries.
While elaboration of underlying principles and generic recommendations are important, we have tried in this document to move beyond that where it seemed possible to do so, and make more concrete, pragmatic recommendations—for instance about consent structure, the methods required to properly prepare data for reuse or the content of data use agreements. We have also identified areas where more exploratory and preparatory work needs to be done, for example in developing quality standards for data repositories that hold clinical research data, or in the need to establish metadata systems and infrastructure to support object discovery. A priority issue within future discussions must be how to ensure the sustainability and financial support of an IPD sharing infrastructure in the long-term, as it was not possible to identify a definitive answer or model at this stage.
We have tried to ensure that the perspective and concerns of the researcher, whether generating data as a trialist or as a secondary data user, have been incorporated into the recommendations. Thus, we have emphasised the need to develop appropriate support systems for planning data sharing and for preparing data, and for finding and accessing the data in ways that respect the concerns of both generators and secondary users.
The future role of repositories
Several questions for the future concern data repositories. These have been recognised as useful tools in allowing data sharing in other scientific domains, and we urge their further development (see principle 9), but so far they have been little used for clinical trial data. The environmental scan performed within the context of this project has shown that there are several repositories already available (eg, B2SHARE, EASY, Zenodo, Dryad, Figshare) that do include at least some clinical trial data sets, and several more are under development (eg, the MRCT’s Vivli). The origin, scope, policies and capabilities of existing repositories are extremely heterogeneous, however, and it is not always clear how their business models will guarantee their long-term sustainability, or what will emerge as the most appropriate organisational model.
For instance, should the research community work towards fewer, larger repositories open to all types of clinical trial data, or is it better served by a smaller number of specialist data stores, perhaps managed by the research communities that are generating the data? If a multiplicity of repositories is inevitable, as more individual institutions, and perhaps countries, establish their own data repositories, how can we make procedures and processes more consistent between them, and confederate content—at least at the metadata level—to make it easier (and cheaper) for those trying to discover content?
A portal supporting identification of trial data stored in repositories, and providing information on access to that data, would make this information more discoverable and would likely increase the reuse of data. Existing approaches to characterising repositories (eg, re3data) should be explored for suitability in the clinical research domain and perhaps adapted or extended. Finally, how should the repositories, whatever their size, be assessed for compliance with standards of good practice, how can that assessment process be financially supported, and how can the results of the assessment be transmitted back to data generators and users?
The need for empirical research
The number of empirical studies about data sharing is, so far, relatively small. A limited amount of data is available, dealing with individual aspects of data sharing (eg, surveys about attitudes and experiences,81 82 statistics about data requests and shared data,83 84 and studies of the costs of data preparation85). Given that the principles and recommendations in our document (and similar reports) are largely consensus-based, further evidence gathering should be a priority. The topics that will need investigation or ongoing monitoring include the following:
The levels of IPD and document sharing, including when, how and why data are made available for sharing, and the differences between planned and actual data sharing activity.
The future levels of IPD and document access requests, and the reasons for those requests.
The costs and time involved in preparing data for sharing, and methods of reducing these.
The attitudes of different stakeholders (researchers, funders, patients, publishers, the general public and others) to IPD sharing and reuse, including the reasons why some people were not making data available in timely fashion.
The incidence and nature of any misuse of information or incorrect secondary analysis, not least because this is a reason often given for reluctance to share data.
The types and quality of research outputs generated from the reuse of IPD, to highlight the value of data sharing.
Comparisons of different access regimes (eg, open, free platform vs controlled access) in terms of costs, accessibility, usage, user feedback and others.
Comparisons of the utility of different data types, specifically anonymised versus pseudonymised data.
Comparisons of different repository systems, including costs, data content and compliance with standards.
Much of this work will be through traditionally funded and published research, examining particular aspects of IPD reuse. Some may examine the impact of specific sharing initiatives, for example, the 2016 SPRINT data analysis challenge organised by the New England Journal of Medicine.86 87 But in some cases (eg, monitoring the outputs generated from reuse, or collating the data about available repositories and the services they offer), it would be more useful to construct continuous monitoring and reporting mechanisms. Some work of this sort is already being carried out, for example by the IMPACT (IMProving Access to Clinical Trials data) Observatory,88 but funding mechanisms need to be developed so that it can be expanded as data sharing grows.
It will also be vitally important to provide patient groups and their representatives with this empirical data, so that they can remain fully involved in future debates on data sharing, and continue to input their perspective on the reuse of data.89
The need for standards and a global perspective
One of the recurrent themes in the current document is the need for standards and standardised processes: for instance, for data and metadata, for repositories, for ways of de-identifying data, for processing request applications and for data use agreements. The use of standards is seen as critical in reducing costs and increasing confidence in the systems and data in use, and it will therefore be important that non-commercial researchers involve themselves in the continuing development of standards of all types. It is also important that standards and standard processes are as global as possible.
Data sharing is, intrinsically, like science in general, an activity with global scope. A global perspective is therefore the best way in which to develop efficient and effective standards, processes and systems. We appreciate that is easy to say but often very difficult to implement, not least because very few funds, outside of United Nation agencies, are made available on a global basis. The alternative, however, of developing national or regional solutions and then attempting to join them up is likely to lead to even more difficulties, and costs, in the long term.
We believe the 10 principles outlined in this report are relevant globally, but we accept that some of the recommendations may not be completely applicable to other contexts (or countries) without adaptation. The recommendations were generated using, in the main, a non-commercial European perspective, with a focus on clinical trials. It will be important to try and explore further how differences in regulations or research systems in countries outside Europe could affect the applicability of these recommendations.
For example, in the USA, recent guidelines have indicated that the sharing of de-identified IPD from clinical trials does not require separate consent from trial participants, assuming that the term ‘de-identified data’ means data that would not constitute identifiable private information in the hands of a third party. Under certain circumstances this ruling can also apply to data released with a code in place (ie, pseudonymous data).90 This is contrary to the position in Europe.
An additional difficulty is that the legislative and regulatory context in many places is rapidly changing. This is the case in Europe, with the introduction of the new General Data Protection and Clinical Trials Regulations, but also (for instance) in Japan, where the Personal Information Protection Act, Clinical Research Act and Next-Generation Medical Base Act were established in March and April 2017. These acts describe how to handle IPD for data sharing and deal with, among other things, informed consent regulations (K Takenouchi and D Nakatani, personal communication, 2017). We have to develop mechanisms to monitor and interpret the changing legislative and regulatory frameworks, and design systems around them appropriately.
We believe that the international task force has constructed a comprehensive framework of policies and procedures for data sharing in clinical trials. The next steps will be to disseminate the principles and recommendations in this framework, engaging different communities and countries, liaising with other major initiatives in the field at regional and global levels, and discuss how the various components of the data sharing infrastructure we need can be funded and implemented.
Supplementary file 1
Acknowledgments
The core team wish to thank the Cochrane IPD Meta-analysis Methods Groups for providing us with useful insights and feedback on the consensus document.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.
Footnotes
Contributors CO, RB, SC, SeB and MM were members of the core group. The core group’s responsibilities were to establish the multistakeholder taskforce, draft intermediate versions of this report, organise and manage the consensus workshops, coordinate the subgroups, and release the final version of the report and paper. HS acted as independent facilitator of the consensus process, chaired the face-to-face meetings together with CO and was responsible for the methods section of the paper. CA, LB, BB, SaB, LC, MD, CD, HF, MF, JG, DG, CG, TG, PH, GK, DK, RLK, KK-J, CK, WK, RK, AL, PSM, AN, JO, PR, IS, DaS, HS, DyS, CT-S, PvR, E-BvV, GRV and JW were members of the multistakeholder task force, attended at least one of the consensus meetings, provided written feedback during the consensus process to draft manuscripts, and approved the final manuscript. JD-M attended all consensus meetings, was responsible for alignment of the work with the H2020-CORBEL project and approved the final manuscript.
Funding This project has received funding from the European Union’s Horizon 2020 research and innovation programme (CORBEL, under grant agreement n° 654248).
Disclaimer The views expressed by MD in this article are the personal views of the author and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties.
Competing interests TG is the editor-in-chief of BMJ Open, the journal that publishes this article. During the paper evaluation she recused herself from the peer review and decision-making process. BB reports various unrestricted gifts (see) supporting travel and effort; grants from Laura and John Arnold Foundation and the Greenwall Foundation during the conduct of the study; and non-financial support from Vivli, outside the submitted work. RK reports she was Founder of CDISC and President during the development of the submitted work. DaS was employed by BioMed Central at the time of the consensus process.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No further data available to share.