Article Text

Abstract
Introduction Transvaginal mesh devices are approved in the USA by the Food and Drug Administration (FDA), through the 510(k) system. However, there is uncertainty about the benefit to harm balance of mesh approved for pelvic organ prolapse. We, therefore, assessed the evidence at the time of approval for transvaginal mesh products and the impact of safety studies the FDA mandated in 2012 because of emerging harms.
Methods We used FDA databases to determine the evidence for approval of transvaginal mesh. To create a ‘family tree’ of device equivalence, we used the 510(k) regulatory approval of the 1985 Mersilene Mesh (Ethicon) and the 1996 ProteGen Sling (Boston Scientific), searched for all subsequently related device approvals, and for the first published randomised trial evidence. We assessed compliance with all FDA 522 orders issued in 2012 requiring postmarketing surveillance studies.
Results We found 61 devices whose approval ultimately relied on claimed equivalence to the Mersilene Mesh and the ProteGen Sling. We found no clinical trials evidence for these 61 devices at the time of approval. Publication of randomised clinical trials occurred at a median of 5 years after device approval (range 1–14 years). Analysis of 119 FDA 522 orders revealed that in 79 (66%) the manufacturer ceased market distribution of the device, and in 26 (22%) the manufacturer had changed the indication. Only seven studies (six cohorts and new randomised controlled trial) covering 11 orders were recruiting participants (none had reported outcomes).
Conclusions Transvaginal mesh products for pelvic organ prolapse have been approved on the basis of weak evidence over the last 20 years. Devices have inherited approval status from a few products. A publicly accessible registry of licensed invasive devices, with details of marketing status and linked evidence, should be created and maintained at the time of approval.
- regulatory approval
- transvaginal mesh
- fda
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
We searched a variety of Food and Drug Administration (FDA) summary database providing listings of all FDA 510(k) device clearances since 1976.
We identified all FDA-522 postmarketing surveillance orders for mesh products issued in 2012.
Searching the latest Cochrane review on Mesh allowed data to be extracted from all of the published randomised controlled trials comparing transvaginal grafts versus traditional native tissue repair in women with vaginal prolapse.
Although we searched extensively for mesh-approved devices, we will have failed to identify some products, especially those that may have been withdrawn from the market.
The lack of a publicly accessible registry of licensed invasive devices with details of marketing status and linked evidence prevents accurate assessment of current status for many implantable devices.
We were unable to scrutinise the European approval system due to its lack of accessibility. The lack of European approval evidence prevents meaningful comparison between the European and US regulatory systems.
Introduction
Implantable devices, such as non-absorbable polypropylene mesh (‘mesh’), are used for surgical treatment of pelvic prolapse and stress urinary incontinence. While evidence suggests that mid-urethral slings are safe and effective,1 2 there have been concerns about the safety of transvaginal mesh used for managing pelvic organ prolapse.3 4 A Cochrane review concluded that although permanent transvaginal mesh may reduce the rates of recurrent prolapse, its use is associated with higher rates of reoperation and bladder damage.5 In 2011, the Food and Drug Administration (FDA) raised concerns about the safety of some transvaginal mesh products.6 Serious adverse events attributed to the use of such products were not rare and included serious complications, such as vaginal erosions, infections and organ perforation.
Implantable devices are approved by the FDA, depending on the class of device. Class II devices are generally approved under the 510(k) process while Class III devices are subject to a Premarket Approval Application (PMA) process. Transvaginal mesh devices were originally included in Class II and required the 510(k) process, but they were reclassified in January 2016. In the 510(k) process, anyone who intends to market a new medical device has to submit a premarketing notification to the FDA at least 90 days before the date scheduled for marketing to begin.7 The regulatory system for medical devices has a low threshold for approval and the limited requirement for clinical data, for which it has been criticised.8 9 The PMA process10 imposes a higher standard of evidence for new devices, including clinical trials. However, Section 510(k) of the Food, Drug and Cosmetic Act allows manufacturers to bypass this requirement by seeking approval for new devices because they are ‘substantially equivalent’ to other devices (referred to as predicates) that are already available.11
Section 522 of the Food, Drug and Cosmetic Act gives the FDA the authority to mandate manufacturers to undertake postmarket surveillance studies of Class II or III devices, among other criteria, when ‘failure would be reasonably likely to have serious adverse health consequences … or the device is to be implanted in the body for more than one year’.12 However, when the FDA asked manufacturers of surgical mesh products to conduct new safety studies in January 2012,13 some manufacturers removed their products from the market, thus preventing the proper assessment of the benefit to harm balance of their devices.14
We set out to systematically assess the nature and quality of the evidence that was available for transvaginal mesh products at the time of their approval by the FDA, and the rate of compliance with orders requiring manufacturers to generate better evidence in response to concerns about harms after approval. We also set out to characterise how assumptions of device equivalence have been used during approvals by mapping the network of devices approved on the basis of equivalence to earlier devices, back to one pivotal device withdrawn from the market two decades ago.
Methods
Approvals and device equivalence
To assess the extent to which device equivalence underpins approval of modern mesh products, we used the 510(k) regulatory approval (K851086) of the 1985 Mersilene Mesh (Ethicon) and the (K963226) 1996 ProteGen Sling (Boston Scientific) to search for related device approvals. We used the FDA summary database for 510(k) Premarket Notification,15 the 510 k.directory/search directory16 and fdazilla.com to search for approvals.17 In combination, these searchable databases provided complete listings of all FDA 510(k) device clearances since 1976, with image PDFs converted into searchable documents using Optical Character Recognition. They also facilitated forward and backward navigation of cross-referenced device applications. Once we found a device in the tree, we then searched for further approvals of that device using the 510(k) number. We developed a ‘family tree of equivalence’ for devices that included the 510(k) number, the year of approval and the current status of the device. We also searched the 510(k) applications using 510kdecisions.com,18 which contains >132000 FDA 510(k) applications submitted since May 1976 for relevant clinical trial evidence for that application.
Randomised trials evidence in the year of approval
We extracted the year of publication from the first published randomised trials for a vaginal mesh product that was included in the Cochrane review published on February 2016.5 This review evaluated 37 randomised controlled trials (RCTs) comparing transvaginal grafts versus traditional native tissue repair in women with vaginal prolapse. We then located the relevant trial device 510(k) application and extracted its year of approval to establish the time from approval to trial publication.
Postmarketing surveillance studies: 522 orders analysis
We identified all FDA-522 postmarketing surveillance orders for mesh products issued in 2012.12 These were obtained from the FDA’s searchable resource19 containing current information on the status of all 522 postmarket surveillance studies. All 522 orders issued in 2012 were obtained, and from these, we extracted all mesh devices named the 510(k) orders, and the current status of the orders, whether active or inactive. We further extracted the reasons underlying the inactive orders and the amount of evidence about the active orders and their reporting status (on the fifth of every month the FDA website posts revised data on interim or final reports).12
Patient involvement
The ideas and design of this study have been formed through long-standing discussions with members of the public, those involved in litigation and members of the media.
Results
Approvals and device equivalence
We found 61 devices related to the 1996 approval of the ProteGen Sling (Boston Scientific; K963226, 1996) and Ethicon’s Mersilene Mesh, which has been used for treating hernias since 1954 (Ethicon K851086, 1985). Figure 1 shows the chain of approvals for these two devices. Searching for the ProteGen ID number K963226k retrieved 22 associated clearances. As examples, these included the 1997 Influence IN-SLING (K972651)20 and the 1998 Tension Free Transvaginal tape (K974098),21 which generated the forward chain.
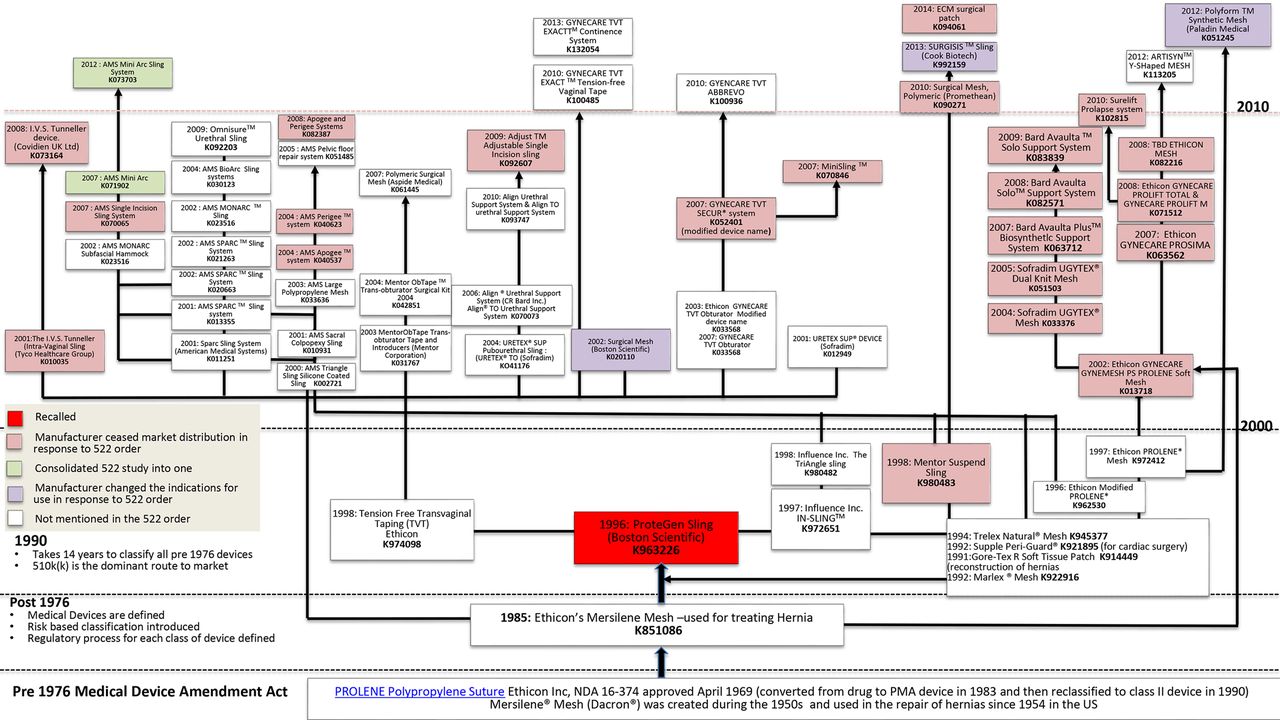
{kind=link}
Food and Drug Administration device chain approval for transvaginal mesh.
Figure 1 also shows that the chain of device approvals using ‘substantial equivalence’ can be traced back through more than one predicate, providing a confusing picture. Newer devices are compared with various older devices, and devices with similar names are compared with each other. For example, the 2008: Avaulta Solo Support System K08257122 has been compared with the 2009: Avaulta Solo Support System K083839, part of the same system.23 Some devices have been removed from the market (eg, 2008: IVS. Tunneller device, Covidien UK; K073164),24 and some have had changes to their indications over time (eg, 2002: Surgical Mesh, Boston Scientific; K020110).25 26
Randomised trials evidence in the year of approval
We found no clinical trial evidence for any of the devices shown in figure 1 in the 510(k) submissions at the time of approval. Table 1 shows that we were able to trace the year of approval and the relevant approval documentation for 29 of 33 different mesh products. In no case was the trial published before approval, and publication occurred at a median of 5 years after approval, range 1 (Gynecare Prolift Ethicon)27 to 14 years (Sofradim, Parieten).28
Comparison of randomised controlled trial publication date of transvaginal mesh and date of US FDA approval
Postmarketing surveillance studies: 522 orders analysis
From the FDA’s website,19 we retrieved 119 orders in 2012 relating to the manufacturers of 126 different vaginal mesh devices, marketed by 34 different companies, 23 of which marketed at least two devices (Search date March 2017). Four companies covered 43% of the orders (American Medical Systems, Boston Scientific, Coloplast Corporation and Synovis Surgical Innovations/Baxter) (see online supplementary web table).
Supplementary file 1
Analysis of the 119 orders showed that in 79 (66%) the manufacturer ceased market distribution of the device subject to the postmarketing surveillance order (if the manufacturer resumes device distribution, they will be required to address the postmarketing surveillance order); in 26 (22%) the 522 orders no longer applied (the indication for use had been revised and no longer included the indication subject of the 522 orders); for 2 (1.7%) orders the manufacturer (Shellhigh) reported that it was no longer in business; and 1 order reported that the device was not a vaginal mesh product. Only 11 orders (9.4%) reported that they were undertaking postmarketing studies. In four orders. the manufacturer requested that their multiple orders should be consolidated into one, leaving a total of seven studies (six cohorts and one RCT) including 2756 participants recruiting (none had reported outcomes).
Discussion
Summary
Our analysis shows a consistent pattern of device approval through the use of ‘substantial equivalence’, with chains of equivalence that ultimately lead back to the Mersilene Mesh. Clinical trials evidence did not form part of the approval process, and when trials were conducted, they occurred a considerable time later (up to 14 years). After the FDA 522 orders requiring postmarketing studies were issued, many manufacturers removed their devices from the market without performing the required research; only seven studies were performed at that stage, and none reported outcomes. Thus, in most cases, there was no opportunity to determine whether the marketed devices had a favourable benefit to harm balance.
Limitations
Though we searched extensively for meshes approved through the FDA 510(k) process, we have failed to identify some of the products, especially those that may have been withdrawn from the market. The withdrawal of some mesh products from the market prevents us from precisely assessing the quality of the evidence for the clinical effectiveness of such products. The failure to identify any clinical trials before regulatory approval of transvaginal meshes casts some doubts on the criteria used by the FDA to grant marketing authorisations for the products; this is further compounded by the lack of postmarketing surveillance for most of the approved products. It is therefore highly likely that more devices could be found in the chain of equivalence shown in figure 1.
Over 100 devices manufactured by at least 40 different companies have at some point been in circulation. A real-time database is needed to allow patients, clinicians and regulators the opportunity to view, and understand, the regulatory chain of any device alongside the evidence used at the time of its approval. We have focused on the USA, which is the only regulatory system that provides transparent access to approvals (called ‘clearances’). In the European Union, approval is overseen by Notified Bodies and is a closed access system29; therefore, it is impossible to scrutinise the European approval system, given its lack of accessibility. The lack of evidence to assess devices approved in Europe prevents meaningful comparison between the European Medicines Agency and FDA regulatory systems.
Findings in context
A US study of devices cleared through the 510(k) process in 2008 found 1105 predicates for 43 new implantable devices. Only eight devices demonstrated evidence to support substantial equivalence of previously cleared devices: most studies provided no data, highlighting the lack of publicly available evidence.9 30 A 2011 analysis of 113 devices recalled because of life-threatening harms showed that recalled Class III devices were often approved using the less burdensome 510(k) process.31 In February 2012, partly in response to problems with transvaginal mesh devices and to address the risk of harms, the US Congress introduced a bill that aimed to alter the 510(k) approval process, and in January 2016 the FDA changed the approval requirements for surgical mesh from Class II to the higher risk Class III. Some mesh devices have been documented as having been marketed in the USA without approval: for example, Johnson & Johnson brought the Gynecare Prolift (K071512) to market in 2005, 3 years before it received regulatory approval, as reported by Bloomberg.32 A previous family tree of the type we present included 16 products.33
Implications
In the USA, the Medical Device Amendment of 1976 decreed that the FDA would review all existing medical devices and put them into one of three classes (Class I, lowest risk; Class II, moderate risk; and Class III, highest risk).34 Once existing devices were classified, all new devices for use in humans were to be labelled as Class III, requiring PMA, which can require evidence from well-controlled investigations (but can also be granted on the basis of lower-quality evidence).35 As the classification of existing devices took >14 years to complete, legislation allowed mesh devices that were developed after 1976 to be approved without the need for clinical data. Consequently, the higher standards required by the PreMarket Approval process for Class III devices were in fact not applied, and mesh devices have consistently used the ‘substantially equivalent’ claim to ensure market access through the less burdensome 510(k) route.
While the PMA route requires clinical data, 510(k) focuses primarily on mechanical performance and material safety; requiring bench and/or animal testing to confirm that certain technical specifications are met, and evidence that mesh is biocompatible. However, the 510(k) regulation allows approval to be obtained by comparison with a chain of equivalent devices, rather than through well-designed clinical trials, which would be preferable.
In response to FDA 522 orders, manufacturers have withdrawn from the market a substantial number of lifelong implantable devices that had not been subjected to clinical trials, leaving uncertainties about their relative benefits and harms. Devices are not subjected to the well-tested methods that have for many years been applied to medicinal products, and this has important implications. For example, a device, the Exhale Drug-Eluting Stent, which appeared to be effective in a phase II study, was found to be ineffective and harmful in phase III.36 37 Furthermore, the FDA has recently reported 22 case studies of drugs, vaccines and medical devices for which promising phase II clinical trial results were not confirmed in phase III studies.38 Better designed and adequately powered trials are required to allow for better assessment of the benefit to harm balance of transvaginal meshes in those most likely to gain some benefit for the repair of pelvic organ prolapse.
Transvaginal mesh for pelvic organ prolapse has not even generally been subjected to adequate clinical studies at any phase of its development. The available trials have been poorly designed and poorly conducted and have failed to account for variable lengths of patient follow-up, underlining the importance of properly designed clinical trials during the development of devices and careful postmarketing surveillance. The IDEAL collaboration has proposed a framework for life-cycle product evaluation whereby devices could be tested before and after marketing.38 39 One such study trialling the benefits and harms of transvaginal mesh for pelvic organ prolapse based on the IDEAL framework was recently published.40
In 2016, device manufacturers, the FDA and professional organisations established the Pelvic Floor Disorders Registry.41 Postmarketing studies and registries of patients treated with such devices should be mandatory at the time of approval.
Conclusions
Our findings highlight the lack of evidence used for licensing transvaginal mesh products. When evidence has been forthcoming, it has often emerged too late to inform clinical practice. The current systems for ensuring patient safety are inadequate for medical devices. We think that clinical trials evidence should be mandatory for marketing authorisation of implantable devices and that there should be incentives for the development of high-quality evidence-based devices. A publicly accessible registry of licensed invasive devices with details of marketing status and linked evidence should be created and maintained.
References
Footnotes
Contributors CJH designed the study, collected the data and drafted the first version for the manuscript. IO, BG, AP, JKA, TJ and KRM contributed to the data interpretation and development of the manuscript and approved the final draft. CJH is the guarantor.
Disclaimer The views expressed are those of the authors.
Competing interests CJH is an expert witness in a current medical device legal case and has received financial remuneration from an asbestos legal case. He has received expenses in the past from the US FDA to attend a working group on device regulation. CJH and BG receive funding from the National Institute of Health Research (NIHR) School of Primary Care. BG receives funding from the Laura and John Arnold Foundation and reports personal fees from intermittent additional personal income from speaking and writing for lay audiences on problems in science and medicine including regulatory shortcomings. TJ and CJH were recipients of a UK National Institute for Health Research grant for a Cochrane review of neuraminidase inhibitors for preventing and treating influenza. Also, TJ receives royalties from his books published by Il Pensiero Scientifico Editore, Rome and Blackwells. TJ is occasionally interviewed by market research companies about phase I or II pharmaceutical products. In 2011-2013, TJ acted as an expert witness in a litigation case related to the antiviral oseltamivir, in two litigation cases on potential vaccine-related damage, and in a labour case on influenza vaccines in healthcare workers in Canada. He has acted as a consultant for Roche (1997-1999), GSK (2001-2002), Sanofi-Synthelabo (2003) and IMS Health (2013), and in 2014 was retained as a scientific adviser to a legal team acting on oseltamivir. In 2014-2016 TJ was a member of three advisory boards for Boehringer Ingelheim and is the holder of a Cochrane Methods Innovations Fund grant to develop guidance on the use of regulatory data in Cochrane reviews. In 2016-2017 TJ was a member of an independent data monitoring committee for a Sanofi Pasteur clinical trial on an influenza vaccine. JKA has published papers and edited textbooks on adverse drug reactions; he has also acted as an expert witness in cases related to adverse drug reactions.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement A spreadsheet recording the individual 522 orders is available on request from the corresponding author.