Article Text

Abstract
Introduction Sepsis is an acute illness resulting from infection and the host immune response. Early identification of individuals at risk of developing life-threatening severe sepsis could enable early triage and treatment, and improve outcomes. Currently available biomarkers have poor predictive value for predicting subsequent clinical course in patients with suspected infection. Circulating leucocytes provide readily accessible tissues that reflect many aspects of the complex immune responses described in sepsis. We hypothesise that measuring cellular markers of immune responses by flow cytometry will enable early identification of infected patients at risk of adverse outcomes. We aim to characterise leucocyte surface markers (biomarkers) and their abnormalities in a population of patients presenting to the hospital emergency department with suspected sepsis, and explore their ability to predict subsequent clinical course.
Methods and analysis We will conduct a prospective, multicentre, clinical, exploratory, cohort observational study. To answer our study question, 3 patient populations will be studied. First, patients with suspected sepsis from the emergency department (n=300). To assess performance characteristics of potential tests, critically ill patients with established sepsis, and age and gender matched patients without suspicion of infection requiring hospital admission (both n=100) will be recruited as comparator populations. In all 3 groups, we plan to assess circulating biomarker profiles using flow cytometry. We will select candidate biomarkers by cross-cohort comparison, and then explore their predictive value for clinical outcomes within the cohort with suspected sepsis.
Ethics and dissemination The study will be carried out based on the principles in the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice. Ethics approval has been granted from the Scotland A Research Ethics Committee (REC) and Oxford C REC. On conclusion of this study, the results will be disseminated via peer-reviewed journals.
Trial registration number NCT02188992; Pre-results.
- ACCIDENT & EMERGENCY MEDICINE
- Sepsis
- Biomarker
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This multi-centre study aims to address an important question in the field of sepsis in the development of a novel predictive biomarker, where our current strategies to identify the deteriorating patient early are poor.
Designed by clinicians in conjunction with immunologists, statisticians, flow cytometry technologists, with industry collaboration, to ensure a robust study design.
Multiple cohort groups for comparison of clinical and immune phenotypes.
Derivation study – any findings will have to be further validated.
Complex study procedures, due to the involvement of clinical and laboratory teams over multiple sites, which will need to be streamlined to develop a clinically useful test for non-tertiary non-urban medical settings.
Introduction
Acute illness secondary to suspected or proven infections account for between 6 and 16% of emergency department (ED) cases.1–3 Approximately half of these patients will have sepsis,2 ,3 defined as infection with signs of systemic inflammation.4 ,5 It has been estimated that around 25% of patients with sepsis syndrome develop severe sepsis (sepsis with organ failure),1 ,6 and 5–10% septic shock (sepsis with cardiovascular failure).1 Mortality increases progressively with these syndromes, with septic shock associated with a 40% mortality.7
Predicting patients with sepsis who are likely to deteriorate is clinically useful, because it could assist clinical triage and decision-making, and result in earlier interventions to prevent organ failure and morbidity. Circulating leucocytes mediate many aspects of the inflammatory response to infection, are easily accessible through blood sampling, and are a potential target for biomarker discovery for predictors of subsequent clinical deterioration.8 Most previous research has focused on soluble mediators, or small studies of immune cell function in a range of sepsis populations.9–13 We hypothesised that measuring leucocyte cell surface markers (hereafter ‘leucocyte biomarkers’) of immune responses by flow cytometry will enable the early identification of infected patients at risk of development of sepsis-associated organ dysfunction or death. We also hypothesised that leucocyte biomarkers will have better discriminant ability than existing soluble inflammation markers that are widely used in clinical practice, such as C reactive protein (CRP) and procalcitonin (PCT). We describe the protocol for a discovery study to explore whether leucocyte biomarkers have potential predictive value for relevant clinical outcomes in patients with sepsis presenting to the ED.
Rationale for study
Cells of the innate immune system (monocytes, granulocytes and dendritic cells) and cells of the lymphocyte lineage (such as T cells, B cells and natural killer cells) have all been reported to have major abnormalities in animal models of sepsis and in selected human studies.8 Broadly, neutrophil life span is prolonged, with immature forms of neutrophils appearing in the peripheral circulation. Monocyte function is impaired, in particular the cell's ability to present antigens alongside downregulation of HLA-DR expression on cell surfaces. In patients with sepsis, lymphocytes are activated, with concurrent increase in inhibitory cell surface molecules and inhibitory cell populations, all of which have been associated with increased risk of secondary infections and death.8 However, most studies have sampled a critically ill sepsis population to characterise these abnormalities; these patients typically have established organ dysfunction and are self-selected as patients developing severe disease. Conceptually, infection leading to a host response resulting in sepsis is a temporal event.14 For these biological processes to be potentially useful as predictive and/or diagnostic tests, they need to be associated with the development of subsequent severe disease in a patient population with acute illness and infection when measured at an earlier time point in the illness trajectory. Specifically, the predictive value for subsequent clinical deterioration is most relevant before significant organ dysfunction has developed.
A potential approach to exploring the association between leucocyte biomarkers and clinical outcomes is to compare the leucocyte phenotypes in a population fulfilling clinical criteria for sepsis but without severe organ dysfunction with a population of patients with established severe sepsis and with matched healthy control populations. This approach has been widely used to provide preliminary evidence for associations between biomarkers and disease conditions, but a major weakness is that patients come from different populations. The gold standard approach to biomarker and diagnostic test discovery is to explore predictive value within a single population of interest, in which the event of interest occurs in a subgroup.15 For sepsis research, the optimum approach is to recruit from a well-defined patient cohort, and follow these cases to ascertain which develop organ dysfunction and/or critical illness.
A diagnostic test that predicts deterioration could have clinical value in a population in whom the pretest probability for progressing to severe sepsis is low, as it may influence the intensity of therapy, monitoring and subsequent placement in the hospital (eg, early admission to critical care). Patients presenting to the ED represent a high volume population who frequently fulfil sepsis criteria, have a low rate of subsequent development of significant organ dysfunction and in whom triage decisions can be difficult. The goal of the Early PREdiction of Severe Sepsis (ExPRES-Sepsis) study is to identify potential leucocyte biomarkers with clinically relevant predictive value in an ED population. Our approach is to characterise the expression of leucocyte surface markers of immune response by flow cytometry in three populations: patients presenting to the ED with suspected sepsis, patients with established severe sepsis in the intensive care unit (ICU) and an age/gender matched non-septic control population in the ED. These three cohorts will be used to select promising leucocyte biomarkers that may have predictive value for subsequent clinical deterioration within the ED cohort. Within the ED suspected sepsis cohort, we will follow patients' clinical course to study the predictive value of leucocyte biomarkers for subsequent development of organ dysfunction and other relevant clinical outcomes. We hypothesise that some immune/inflammatory cell surface markers, measured in a population of patients presenting to hospital with sepsis, will have predictive value for the subsequent development of clinically severe disease characterised by the development or worsening of acute organ dysfunction.
Aims
To characterise the expression of leucocyte biomarker abnormalities by flow cytometry in patients presenting with suspected infection, non-infective acute illness and critically ill patients with severe sepsis;
To test the ability of these markers to predict clinical deterioration in patients with suspected infection;
To test the association between these biomarkers and subsequently confirmed infection;
To compare the performance of promising leucocyte biomarkers with established soluble markers of infection (CRP and PCT).
Methods
Study design
ExPRES-Sepsis is a prospective, multicentre, exploratory, cohort observational study.
Study sites
The study will recruit from four sites in the UK: Royal Infirmary of Edinburgh (Edinburgh, Scotland, UK); Western General Hospital (Edinburgh, Scotland, UK); Royal Victoria Infirmary (Newcastle, England, UK); St Thomas' Hospital (London, England, UK).
Definitions
Sepsis is defined as a systemic inflammatory response syndrome (SIRS) due to infection.4 The criteria for SIRS are two or more of the following four physiological responses to a clinical insult: (1) temperature >38°C or <36°C, (2) heart rate >90 bpm, (3) respiratory rate >20 breaths per minute, or arterial carbon dioxide tension <32 mm Hg (equivalent to 4.3 kPa), (4) white cell count >11 or <4 (×109 cells), or 10% immature (band) forms.4 Severe sepsis is sepsis with one of organ dysfunction, hypoperfusion or hypotension.4 Septic shock is the presence of hypotension due to sepsis which persists despite adequate fluid resuscitation.4 Critical care is defined as an ICU or high dependency unit as used in the UK.16 ,17
Diagnosis of sepsis, severe sepsis and septic shock
In the ExPRES-Sepsis study, we will diagnose sepsis in a patient where they meet at least two out of four of the SIRS criteria, plus one of the following: (1) suspected infection being investigated with blood cultures and/or treated with antibiotics, (2) clinically confirmed infection (derived from Horan et al,18 see online supplementary appendix B) or (3) microbiologically confirmed infection (see online supplementary appendix B).
Supplementary appendix
We will diagnose severe sepsis as: (1) the patient has been diagnosed with sepsis, and (2) the patient has organ dysfunction. Organ dysfunction will be measured by the Sepsis-related Organ Failure Assessment (SOFA) score.19
Septic shock will be diagnosed in critical care units, using modified PROWESS-SHOCK trial criteria, where: (1) the patient has been diagnosed with sepsis; (2) the patient has required vasopressors for 4 hours, consisting of either (A) norepinephrine at >5 μg/min, (B) epinephrine at >5 μg/min or (c) vasopressin at >0.03 units/min; and (3) evidence of organ hypoperfusion.20 Organ hypoperfusion is defined as one of the following: (1) base excess of ≥5 mmol/L, (2) bicarbonate of <18 mmol/L, (3) lactate of >2.5 mmol/L, (4) urine output of <0.5 mL/kg/hour, (5) 50% increase in creatinine from baseline, (6) hepatic dysfunction alanine transaminase/aspartate transaminase >500 IU/L or (7) bilirubin >2 mg/dL (34.2 μmol/L).20 ,21
Septic shock will be diagnosed in non-critical care settings where the patient meets ALL of the following criteria: (1) sustained systolic hypotension (systolic blood pressure <90 mm Hg for ≥4 hours), (2) despite adequate fluid resuscitation (≥30 mL/kg of any intravenous fluid) and (3) during the 8 hours following start of the episode of systolic hypotension.20
Patient cohorts
We will recruit three distinct patient cohorts:
Cohort 1: patients presenting to hospital with suspected infection with a systemic inflammation (sepsis; discovery cohort).
Cohort 2: hospitalised patients with community-acquired severe sepsis requiring treatment in critical care (true-positive cohort).
Cohort 3: patients presenting with no suspicion of infection or systemic inflammation, needing hospitalisation (‘non-sepsis comparison population’).
Sample size
Cohorts 1 and 2: The sample size for cohorts 1 and 2 was calculated on a combination of: (1) an expected event rate of 5–10% in cohort 1 of developing the outcome of interest (severe sepsis and/or critical care admission), (2) confidence interval (CI) widths for positive and negative predictive values (PPV and NPV, respectively) and (3) the number of ‘true positives’ required in cohort 2.
In a cohort 1 sample of 300, we expect 15–30 events of interest. With 100 true positives in cohort 2, the numbers of patients with severe sepsis and/or critical care admission in cohorts 1 and 2 (400 patients overall) combined will be 115–130—around 25–30%. Assuming a range of PPV/NPV performance from 50% to 90%, the CI widths would range from ±4.6% to ±9.8% for PPV, and ±3.4% to ±6.3% for NPV. We considered this level of precision acceptable for an exploratory derivation study.
Cohort 3: The non-sepsis population (n=100) will be used as a comparator group to explore candidate markers that have potential predictive value in the population of interest, and was chosen to be equivalent in size to cohort 2. Determining leucocyte biomarkers with little variance in non-septic patients will be an important property in relation to potential utility for detecting sepsis. An a priori-defined sampling strategy was designed, starting midway through the recruitment to cohort 1, to ensure the age and gender profile of cohort 3 matches cohort 1 (table 1).
Cohort 3 age and gender matching matrix
A detailed trial analysis plan will be agreed before the database is locked, and will take into account the clinical characteristics of the populations enrolled in the study, especially event rates and final numbers recruited (see below).
Screening procedure
Screening of consecutive patients will be performed by qualified individuals designated by the local principal investigator (PI). A screening log will be maintained at each site to capture reasons for non-inclusion of eligible patients.
Inclusion criteria
Cohort 1
Age ≥16 years (≥18 years in England), (2) SIRS criteria met, (3) clinical suspicion of sepsis (blood cultures and/or other samples taken for microbial culture, or antibiotics started by clinical team), (4) no clinical suspicion of severe sepsis or septic shock at the time of enrolment and (5) enrolled within 12 hours of hospital (ED) presentation.
Cohort 2
Age ≥16 years (≥18 years in England), (2) SIRS criteria met, (3) clinical suspicion of sepsis (blood cultures and/or other samples taken for microbial culture, or antibiotics started by clinical team), (4) severity of sepsis requiring critical care admission (based on decision of caring clinical teams), (5) enrolled within 72 hours of hospital admission and (6) not enrolled into cohort 1 of ExPRES-Sepsis.
Cohort 3
Age ≥16 years (≥18 years in England), (2) does not meet SIRS criteria, (3) no clinical suspicion of sepsis (blood cultures and/or other samples NOT taken for microbial culture, and antibiotics NOT started by clinical team), (4) patient expected to be admitted to hospital, (5) patient NOT expected to die during hospital admission.
Exclusion criteria (for all cohorts)
Exclusions were chosen to ensure conditions that provoke a sterile inflammatory response or lead to immune dysfunction did not act as confounders during flow cytometry analysis. Patients who would not be actively treated were also excluded.
The exclusion criteria are any of: (1) acute pancreatitis, (2) haematological malignancy, (3) recent chemotherapy (past 2 weeks), (4) myelodysplastic syndromes, (5) known neutropenia, (6) HIV infection, (7) viral hepatitis infection, (8) pregnancy, (9) blood transfusion >4 units in past week, (10) oral corticosteroids for >24 hours prior to enrolment, (11) decision not for active therapy/for palliative care at admission and (12) inability to consent the patient.
Consent
Informed consent is obtained from the patient, or from their relatives when they are incapacitated due to sepsis, for recruitment to ExPRES-Sepsis by trial research nurses. If a patient regains capacity, their personal consent to remain in the study is sought.
Clinical management of patients in the study
Clinical management of patients in this study will remain entirely at the discretion of the treating clinician: no treatment alterations will occur as a result of study participation. Data will be collected to benchmark clinical management against national standards for sepsis management, the ‘sepsis six’, for cohort 1.22 ,23
Study assessments
Recruiting staff will be blinded to the results of laboratory analysis; conversely laboratory staff are blinded to clinical data.
Blood sampling
Up to 10 mL of blood will be taken at each sampling point (table 2) for flow cytometric analysis of leucocyte biomarker expression. Where possible, this will be at time of venous access insertion or sampling for clinical indications. The sequential sampling within cohort 1 will be completed whenever possible for exploratory analyses of leucocyte biomarkers changes over time. Sequential sampling will only occur when the patient remains in hospital, the patient agrees and it is technically feasible.
Early PREdiction of Severe Sepsis (ExPRES-Sepsis) sampling schedule
Further handling of blood samples
Leucocyte surface markers of immune responses will be measured using flow cytometric analysis within 4 hours of sample acquisition. Blood is held at 4°C for processing. Excess blood not used for flow cytometric analysis will be frozen and stored as blood plasma for future analysis, including CRP and PCT.
Flow cytometry data collection
Leucocyte biomarker expression will be recorded using antibody staining and measurement by flow cytometry.24 Antibodies are supplied by Becton Dickinson Biosciences (BDB). All sites use antibodies from the same batch. Antibodies selected for cell surface staining have either shown previous association with differential expression in inflammation, and therefore noted to be of potential in the development of a clinical test, or are used to select leucocyte subtypes of interest. Our aim is to explore a wide range of potentially relevant cell surface biomarkers on different immune cell types and subtypes. Specific assays will be developed for the purpose of the study, and agreed prior to undertaking the analyses in real time during the clinical study. The exact biomarkers chosen are not detailed in this protocol manuscript, in part because of IP issues. The final biomarker panel selected, their reliability and the performance of all those evaluated will be included in the main manuscript (including those rejected due to poor reliability).
All sites have standardised on the same platform, the FACSCanto II (BDB, San Jose, California, USA), for flow cytometric analysis of samples. Machines will be standardised by monthly matching of target values using a common batch of BDB Cytometry Setup and Tracking beads, and daily internal quality control using these beads. Staining, data capture and storage will be conducted in accordance with a single study standard operating procedure.
Flow cytometry data will be held centrally. Final analysis will be conducted using a single analysis platform, VenturiOne (Applied Cytometry, Sheffield, UK).
Clinical data collection
Patients in cohort 1 will be followed to record their subsequent clinical status and specifically for the development of organ dysfunction. Patients in cohorts 2 and 3 will only have data collected at the time of blood sampling.
The following data will be collected from each patient: demographics (age, gender, functional comorbidity index25); Acute Physiology and Chronic Health Evaluation (APACHE) II score on study entry; SOFA score at enrolment, 24 and 72 hours; standardised early warning system (SEWS) score at enrolment, 24 and 72 hours; clinical data (admission diagnosis, length of stay, maximum level of care); treatment data (antibiotics, use of vasopressors, volume of fluid administered during first 12 hours, recording of critical care interventions); final diagnosis; infection data—including confirmation or refutation of diagnosis of infection (see online supplementary appendix B), source of infection and results of microbial cultures; need for source control procedure (surgical operation, percutaneous drainage, endoscopic treatment); ceiling of care decisions (eg, anticipatory care planning against cardiopulmonary resuscitation,26 patient judged not suitable for critical care); and conventional laboratory test results taken as part of standard care.
Analysis plan
Analysis of the data set in ExPRES-Sepsis will follow an iterative process to test, eliminate and validate potential biomarkers and outcomes of interest (figure 1). The flow cytometry and clinical expertise within the investigator group will allow us to optimise the biomarkers that we will select to explore associations with our outcomes of interest in the final stages of analysis.

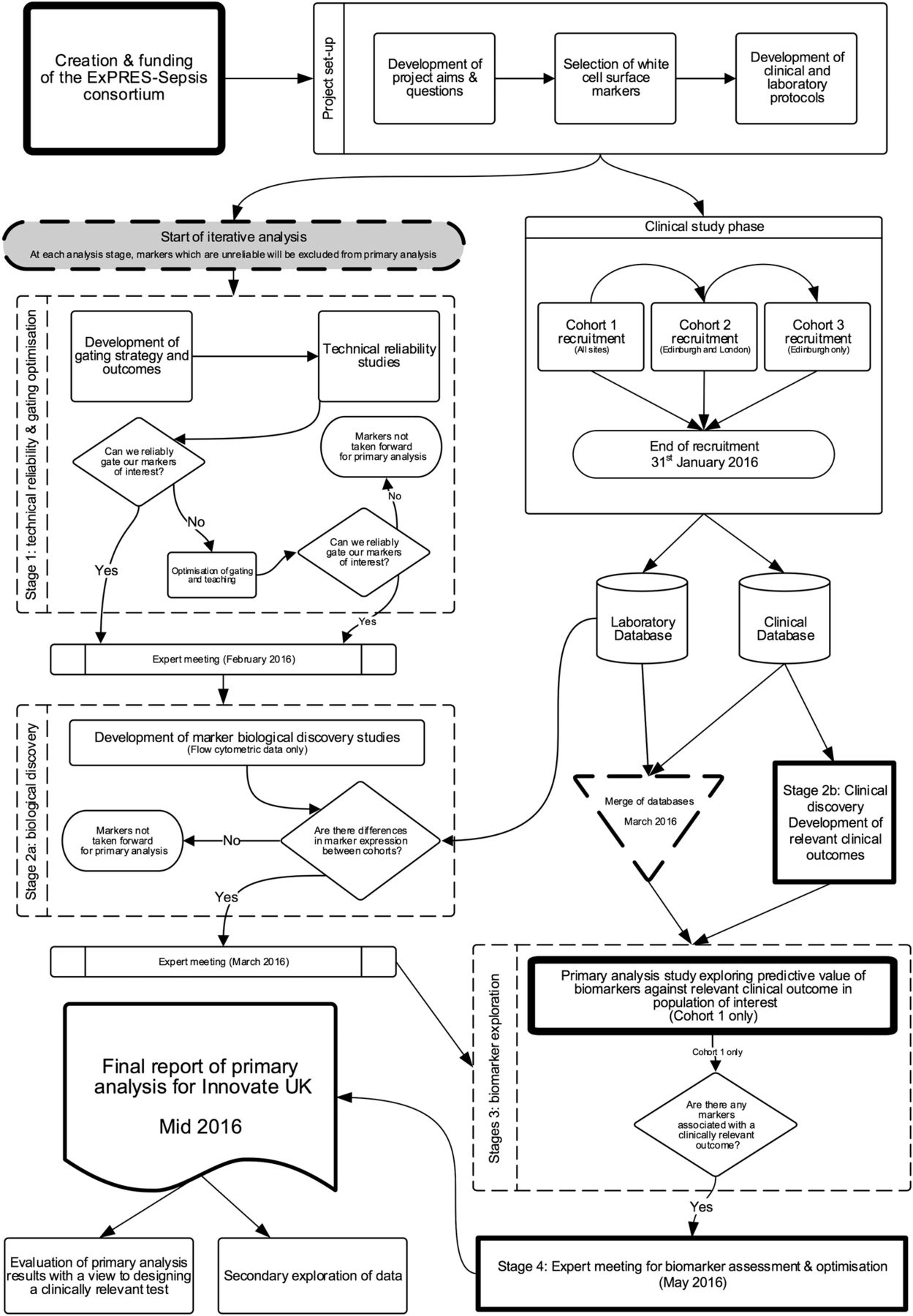{kind=link}
ExPRES-Sepsis analysis flow chart.
Stage 1: reliability and optimisation
The first stage of analysis will use the raw flow cytometry data, without any matching clinical data, from all cohorts to explore whether the flow cytometric readings (gating) of each marker are reproducible.27 Interobserver and intraobserver reliability studies will be conducted with three expert readers of the data. Fifty data files per reader will be analysed to determine interobserver agreement, with 13 files for intraobserver agreement.28 After an initial reliability study, a learning and optimisation stage will occur using expert meetings to refine gating strategies in problematic markers to ensure we do not incorrectly reject markers. Thereafter, a second reliability study, using the same sample size, will be conducted. The results will be reviewed at a further meeting of the research team, including flow cytometry experts, to select markers that show robust reliability to take forward for further analysis.
Stage 2: discovery
Stage 2 has two parallel processes of discovery—biological and clinical. The two data sets (laboratory and clinical) will continue to be kept separate, with researchers blinded to the matched immune and clinical phenotypes of the patients.
Biological discovery
Leucocyte biomarker expression will be compared across all three cohorts, without matching clinical data, to rationalise markers that may be associated with the clinical phenotype of interest, namely severe sepsis with (or without) organ dysfunction. Specifically, we will identify potential leucocyte biomarkers that show differential expression between the three clinical phenotypes. A further expert meeting will take place to review these results in combination with relevant literature relating to each of the leucocyte biomarkers. At this meeting, the reliability data, detailed review of existing literature, results from the cross-cohort analysis of the study and expert consensus will determine the selection of biomarkers to be used for further analysis (see below). The rationale and justification for all inclusion and rejection of candidate biomarkers will be described and documented at this time. The aim will be to select leucocyte biomarkers that have sufficient measurement reliability to be used in clinical practice and show promise for discriminating between the study cohorts, particularly between cohort 2 (established severe sepsis) and cohort 3 (matched non-septic controls).
Definition of clinical outcomes of interest
This phase will be limited to cohort 1, the population of interest in whom we aim to predict outcomes. Several outcomes have been chosen. We justify this by the exploratory nature of the study, and in order to include clinical phenotypes of differing severity based on organ dysfunction and clinical destination at 24–72 hours.
At the time of designing the study, we proposed a primary outcome of septic shock and expected that 5–10% of enrolled patients would develop this during follow-up. During the conduct of the study, in May to June 2015, an interim analysis of the clinical data was performed to explore the event rate for septic shock. This indicated that rates were significantly lower than anticipated prior to the study (<5%). In July 2015, the decision was made to change the main outcome to the development of organ dysfunction during the 24–72 hours following recruitment. This was considered clinically relevant because it is associated with significantly increased mortality and the need for critical care admission in many cases. A diagnostic test that could predict this outcome at the time of assessment in the ED would have potential clinical value. The final decision regarding the cut-off level of organ failure severity for the analyses was made after the publication of the revised international sepsis definitions in March 2016.29 These suggested a SOFA score increase of ≥2 as the cut-off for defining sepsis, with the assumption that the preacute illness SOFA was 0 unless clear evidence of chronic organ dysfunction contributing to SOFA points was available. We therefore chose a SOFA score of ≥2 as main outcome, and a more severe value of ≥4 as a secondary outcome.
The outcomes of interest that will be used in cohort 1 to explore the potential predictive value of the novel leucocyte biomarkers will be:
Primary measure
Sepsis-related Organ Failure Assessment (SOFA) score ≥2 at 24 or 72 hours or both following presentation to hospital.
Secondary measures (in order of priority)
Critical care admission, or death, within 72 hours of presentation;
Organ dysfunction (SOFA≥4) at 24 or 72 hours or both following presentation to hospital;
Discharge home within 72 hours;
Discharge to home, or in hospital with no organ failure, within 72 hours of presentation;
Death from sepsis;
Length of hospital stay;
Confirmed infection (clinical and/or microbiological confirmation);
Microbiologically proven sepsis;
Development of septic shock.
Justification and rationale for outcomes of interest
Higher levels of organ dysfunction, as assessed using SOFA score,19 are associated with higher mortality, morbidity and illness costs. Observational studies show that in critical care populations, there is 9% mortality rate in patients with no organ dysfunction, compared with 22% and 83%, respectively, in patients with one and four organ failures.30 It therefore can be considered a pathophysiological surrogate for ‘critical care admission within 72 hours’; however, the SOFA score will provide more stratified information on the patients' condition, and is consistent with the new definitions of sepsis that emphasise the prognostic importance of organ failure.29
The need for critical care admission within 72 hours of first presentation to hospital is a relevant clinical outcome that identifies patients requiring higher intensity of care, who may benefit from ‘early’ clinical interventions for sepsis and/or early admission for monitoring. Conversely, discharge to home within 72 hours of first presentation to hospital indicates an outcome indicative of low illness severity. These two outcomes represent an important dichotomy in the population when reviewed in the ED. A test that could change the pretest to post-test probability of both outcomes has potential clinical utility as a decision-making tool. A further outcome of discharge to home within 72 hours, or in hospital with no organ failure at 72 hours, is similar to outcome 2, but will also identify the group of patients who would be medically suitable for discharge home, but are prevented from doing so for social or other reasons.
Prediction of microbiologically positive infection lends itself to a clinically useful test. However, this outcome is exploratory as it has been previously shown that many patients with sepsis have negative blood cultures, with only 30–40% positive in severe sepsis/septic shock.11 ,31 ,32
Stage 3: biomarker exploration
Clinical data will be linked to leucocyte biomarker data in this stage of analysis. The leucocyte biomarkers selected in stage 2 will be analysed for their ability to predict the development of the clinical outcomes of interest using receiver operator characteristic (ROC) curves to determine optimum cut-off points. The PPV, NPV, positive and negative likelihood ratio, sensitivity, specificity and area under ROC curve will also be calculated for each marker. Ninety-five per cent CIs will be reported for all measures of predictive accuracy. Comparative performance characteristics for tests used in current routine management of the patients, namely CRP and PCT, will also be calculated.
Stage 4: clinical optimisation
The biomarker performance data will be examined, and further discussed with experts within the research group and external independent experts, to assess the potential clinical utility for the leucocyte biomarkers. At this stage, the potential for combining biomarkers to increase and optimise predictive ability will be explored.
Ethics, regulations and governance
Study management
The study is managed by the Edinburgh Clinical Trials Unit (ECTU) and the Edinburgh Critical Care Research Group at the University of Edinburgh, Scotland. The study sponsor and funders will not have any decision making authority in the conduct of this study once approved.
Sponsorship
The study is co-sponsored by University of Edinburgh and National Health Service (NHS) Lothian.
Duration and current status of study
The study is planned to run for 3 years. The first patient was recruited in January 2014. At the current time (25 January 2016) recruitment to each cohort is ongoing. The expected end date for recruitment is 31 January 2016. We expect to have completed our primary data analysis plan with a view to dissemination of results by August 2016.
Ethical and regulatory approvals
The study will be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and the International Conference on Harmonisation Good Clinical Practice.33 The CI, site PIs and clinical research staff have completed the relevant training with up-to-date certification. Local research and development approval has been granted for each study site. The study has been registered with the UK National Institute for Health Research (NIHR) Clinical Research Portfolio.
Dissemination
The study will be reported according to the STARD guidance for reporting diagnostic accuracy studies.34 The findings will be disseminated via peer-reviewed publications, and at national and international meetings. A lay person’s summary of the principal findings will be sent on request to all patients involved in the study. In addition, the most significant results will be communicated to the public through press releases.
Protocol compliance
The investigators will conduct the study in compliance with the protocol given favourable opinion by the Research Ethics Committee (REC). Protocol changes will require REC favourable opinion prior to implementation, except when modification is needed to eliminate immediate hazards to patients. The ECTU in collaboration with the Sponsor will submit all protocol modifications to the REC for review in accordance with the governing regulations. Protocol compliance will be monitored by the trial manager who will ensure that the trial protocol is adhered to and that necessary paperwork (case report form, patient consent) are being completed appropriately. Any deviations from the protocol will be fully documented in source documentation and case report form.
Patient confidentiality
In order to maintain confidentiality, all case report forms, stored samples and study reports will identify patients by the assigned unique study identifier number only. The only link between the patient's identity and the unique study identifier number will be held securely at the relevant study site.
Summary
The ExPRES-Sepsis study is a large multicentre exploratory study to identify potential novel leucocyte biomarkers that predict subsequent clinical deterioration in sepsis in patients early in their presentation to hospital. The measurement of leucocyte surface markers is a promising strategy for a predictive test. The recruitment and analysis plan ensures we have a robust biomarker set for prediction of clinically relevant outcomes. If we find a positive association with clinical interest, further validation of these markers could lead to the development of a clinically useful early test in sepsis.
Supplementary appendix
References
Footnotes
KAB, TAM and SC are previously affiliated with the institutions listed.
Twitter Follow Deepankar Datta at @deepankardatta, Andrew Conway Morris at @andymoz78 and Manu Shankar-Hari at @msh_manu
Collaborators (see online supplementary appendix A).
Contributors DD, ACM, JA, NW, KAB, JW, AJS, JR, GH, SML, TAM, CJW, ID, JK, AGR, MS-H and TSW were all involved in the design of the study. DD designed the analysis plan and compiled the information for this protocol. JR, GH, SML, TAM and DD are involved in the acquisition and analysis of the data in the study. All authors were involved in the drafting and approval of this protocol. All authors satisfy the four criteria for authorship as per the ICMJE ‘Recommendations for the Conduct, Reporting, Editing, and Publication of Scholarly Work in Medical Journals’ (December 2015).34
Funding This work was supported is InnovateUK as project number 101193, with further funding by Becton Dickinson Biosciences. CJW was also supported in this work by National Health Service (NHS) Lothian via the Edinburgh Health Services Research Unit.
Competing interests All authors' work on this study was funded by InnovateUK, with further support from BD Biosciences. DD reports grants outside the submitted (non-related) work from the Edinburgh and Lothians Health Foundation, and non-financial support from Nova Biomedical. CJW reports support within the submitted work from NHS Lothian via the Edinburgh Health Services Research Unit.
Patient consent Obtained.
Ethics approval Scotland A Research Ethics Committee (REC)—registration 13/SS/0023—and from the Oxford C REC—registration 13/SC/0266.
Provenance and peer review Not commissioned; externally peer reviewed.