Article Text

Abstract
Introduction The efficacy of pharmaceuticals is most often demonstrated by randomised controlled trials (RCTs); however, in some cases, regulatory applications lack RCT evidence.
Objective To investigate the number and type of these approvals over the past 15 years by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA).
Methods Drug approval data were downloaded from the EMA website and the ‘Drugs@FDA’ database for all decisions on pharmaceuticals published from 1 January 1999 to 8 May 2014. The details of eligible applications were extracted, including the therapeutic area, type of approval and review period.
Results Over the period of the study, 76 unique indications were granted without RCT results (44 by the EMA and 60 by the FDA), demonstrating that a substantial number of treatments reach the market without undergoing an RCT. The majority was for haematological malignancies (34), with the next most common areas being oncology (15) and metabolic conditions (15). Of the applications made to both agencies with a comparable data package, the FDA granted more approvals (43/44 vs 35/44) and took less time to review products (8.7 vs 15.5 months). Products reached the market first in the USA in 30 of 34 cases (mean 13.1 months) due to companies making FDA submission before EMA submissions and faster FDA review time.
Discussion Despite the frequency with which approvals are granted without RCT results, there is no systematic monitoring of such treatments to confirm their effectiveness or consistency regarding when this form of evidence is appropriate. We recommend a more open debate on the role of marketing authorisations granted without RCT results, and the development of guidelines on what constitutes an acceptable data package for regulators.
- Regulatory
- CLINICAL PHARMACOLOGY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
This work is the first systematic attempt to identify drugs approved without randomised evidence.
The in-depth review identifies all evidence for treatments throughout the clinical development programme, not just the pivotal study.
Because of the different remits and processes of the European Medicines Agency and the Food and Drug Administration, it is not possible to compare across all disease areas.
Despite identifying treatments without randomised evidence, it is not in the scope of this study to conclude on the appropriateness of approval on the basis of non-randomised data.
Due to the lack of follow-up studies, it is also not possible to reach a conclusion on the efficacy of these products.
Introduction
The efficacy of pharmaceuticals is usually evaluated for regulatory purposes using randomised controlled trials (RCTs) that, when properly designed and conducted, provide unbiased estimates of treatment effect.1 However, there are occasions when a therapy is administered to all patients within a trial; this is a frequent step in the development process of pharmaceuticals. While it is not necessarily a preferred approach, the product can be submitted to regulatory agencies for approval following these studies. This can occur with treatments that are new to the market, or for the extension of a licensed treatment to a different patient population, with the argument that clinical equipoise no longer applies. As the treatment is obviously effective, there is no clinical uncertainty, and as such, to withhold the treatment from one group in a trial would be unethical or impractical.2–4
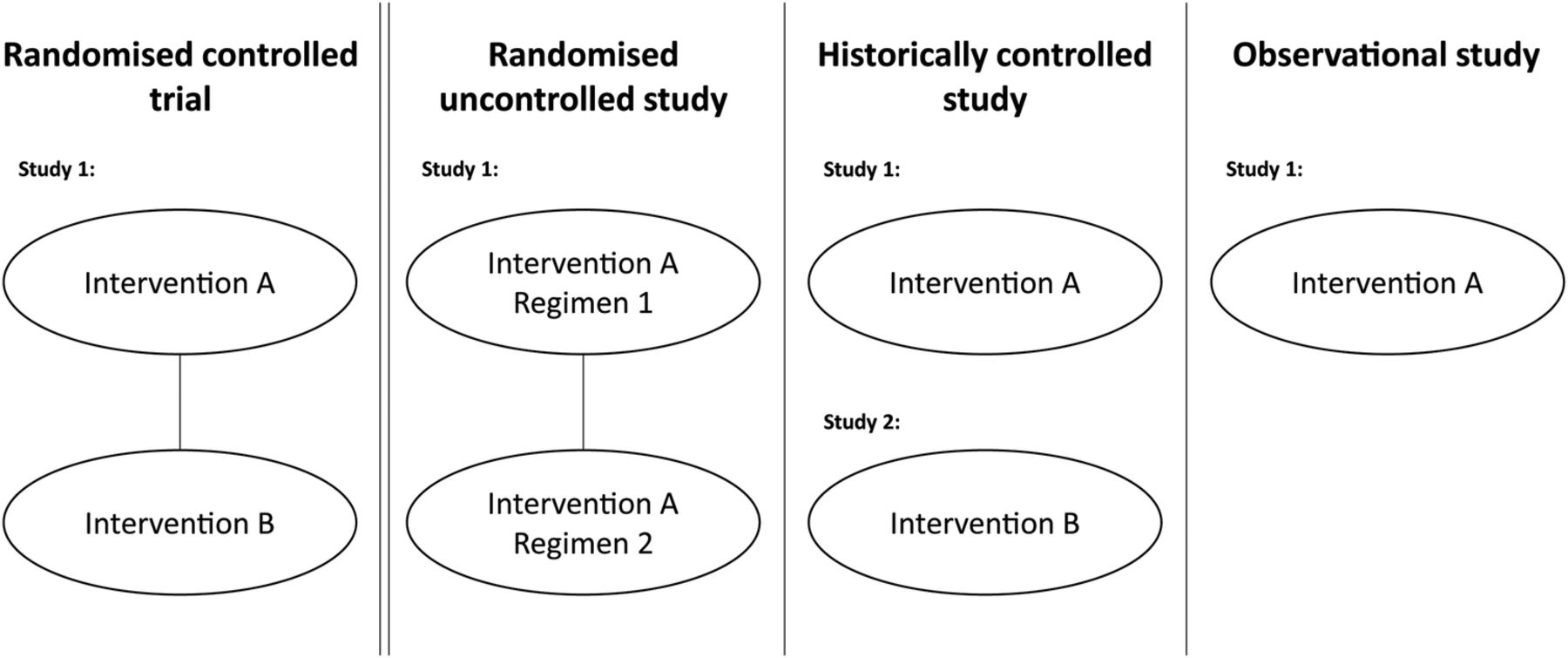
Uncontrolled clinical studies may take several forms that include aspects of randomisation (figure 1). For example, patients may be randomised to different dosages (or regimens) of the experimental treatment. Other data packages may include a clinical study compared to a historical control, such as a previously published study of similar patients treated with the relevant comparator.5 Finally, other treatments may have no control arm or control data, but instead rely on an ‘obvious’ difference, or avoidance of an expected outcome—for example, death from poisoning. The common factor in each of these designs is that the study in isolation is insufficient to demonstrate the effectiveness of the treatment (ie, external data are needed) as it does not contain an internal control arm (either active or placebo) against which outcomes can be compared.

Types of uncontrolled studies used to support regulatory applications compared to randomised controlled trials.
Following the clinical study programme, the data package for a treatment is submitted to the regulator, who must give their approval before a drug is allowed on the market. In the EU and the USA, there are slightly different paths to approval. Historically, pharmaceutical licensing in the EU was the responsibility of individual countries; however, with the development of the centralised authorisation procedure by the European Medicines Agency (EMA) in 1995, companies submit a dossier to the EMA. The EMA reviews the submission, asks questions to the company as a part of the process, and then issues a decision that is binding throughout the EU.6 In the USA, manufacturers submit a New Drug Application (NDA) to the Food and Drug Administration (FDA) which will review the data submitted before making a decision on the request.7
The objective of this study was to review new pharmaceutical approvals by the EMA and the FDA, from 1999 to 2014, to understand how frequently applications (and approvals) are made solely on the basis of uncontrolled studies, without either the pivotal study or supportive studies being RCTs in the indication (previous work has focused solely whether the pivotal study contained a control arm8). A secondary analysis compared the approval rates of the EMA and the FDA for evidence of differences in the approach taken to comparable applications in approval rate and timing.
Methods
Details of the trials conducted for the licensing application of treatments were identified through publicly available regulatory documents. In the case of the EMA, the main sources of information were the European Public Assessment Reports and the Committee of Human Medicinal Products Assessment Reports, while for the FDA, these were the approved label and the Medical Officer's report.
All sources were taken directly from the websites of the EMA and the FDA. The EMA approval database was downloaded on 8 May 2014 (http://www.ema.europa.eu/ema/), and the FDA ‘Drugs@FDA’ database was downloaded on 15 May 2014 (http://www.fda.gov/Drugs/). These databases were used to explore drug approvals based on uncontrolled trial results from 1 January 1999 to 8 May 2014. This date range was chosen as it started several years after the beginning of the EMA Centralised Procedure (1995), giving time for the process to become established. Each approval was reviewed in turn to identify applications made without supportive RCT results.
Following deduplication, all products not approved by both agencies were searched for individually on the website of the other agency with no date restriction to understand whether they were approved before the date period. Searches were also conducted on the website of the manufacturer, and in PubMed and Google News for information regarding submissions, in order to identify any products that did not appear in the databases use for the main analysis.
The aim of our study was to look at newly approved indications for pharmaceuticals, which led to the exclusion of several types of product, listed in table 1. Applications were compared against the exclusion criteria, and if excluded, the reason for exclusion was noted in a hierarchical fashion. If a regulatory application for a product was made to one agency but did not appear in the results for the other agency, a search with no date restriction on was performed to identify any applications made outside the review window (to ensure all comparable approvals have been included).
Exclusion criteria and rationale
The included data were for each approved indication, other approved indications for the treatment, submission and decision dates, disease area and supporting evidence for the approval.
Results
From January 1999 to May 2014, the EMA issued 795 approvals, including 44 indications approved solely on the basis of uncontrolled trials (figure 2). Of the 44 approvals, 8 were extensions of indications for treatments with RCTs in other approved indications, while the remaining 36 approvals were for products in which there were no RCT results in an approved indication. Over the time period, for nine applications without supporting RCT results, the EMA rejected the application or the manufacturer withdrew its submission with a negative recommendation in place.

{kind=link}
{kind=link}
PRISMA diagrams—drug approvals based on uncontrolled clinical studies by the FDA and the EMA.
In the same time period, the FDA issued 774 approvals, which included 60 indications approved solely on the basis of uncontrolled studies (figure 2). Of the 60 approvals, 12 were extensions of indications for treatments with RCTs in other approved indications, while the remaining 48 were for products in which there were no RCT results in an approved indication. Only one application made without controlled trial results (temporfin for head and neck cancer) received was not approved.
Deduplicating for approvals granted in both regions, 74 indications were approved over the period by at least one agency on the basis of uncontrolled trials (table 1). Thirty-four approvals (the largest group) were in haematological oncology, 15 in solid tumour oncology, and 15 in rare metabolic conditions (including enzyme replacement therapies). Outside of these areas, eight approvals were granted in poisoning or emergency medicine and two in general haematology. All the approvals that were extensions of the license for existing therapies were in either haematological oncology or solid tumour oncology (table 2).
Drugs submitted to the EMA and the FDA containing only uncontrolled clinical studies
Searching for treatments approved by only one agency on the basis of only uncontrolled studies without a date restriction yielded further four approvals, which was used in the comparison of EMA and the FDA approval rates and times. Four treatments approved by the EMA 1999 in our date range were approved prior to 1999 by the FDA with a similar data package.
For applications made in both regions, 44 applications were made to both the EMA and the FDA without controlled results (including the 4 made to the FDA prior to 1999), with the EMA approving 35 and the FDA approving 43. Of the 34 applications approved in both regions, the EMA approval was granted a mean of 13.4 months later (median 6.7, IQR 4.5–17.2 months). This delay consisted of two parts: first, companies submitted to the EMA a mean of 7.4 months later (median 1.5, IQR 0.1–8 months), with 28 of 34 indications submitted to the FDA first. Second, the EMA took an average of 6.3 months longer to complete their review and approve products (median 0.2, IQR 0.1–0.3 months); in comparable approvals, the EMA review was quicker in only 3 of 34 applications leading to 30/34 products reaching the market first in the USA.
In addition to the EMA approving fewer products and taking longer to approve products, five treatments were approved with only uncontrolled studies by the FDA, but approved by the EMA at a later date when comparative results were available (a delay in approval of 21.5 months mean/9 months median).
Discussion
The number of approvals without supporting RCT evidence was in excess of what we had expected, with a mean of ∼5 indications per year approved by either (or both) the EMA and the FDA. We had also expected the majority of approvals to be license extensions of existing products; however, this was not the case—only 19% of approvals were license extensions of products demonstrated to be efficacious in RCTs in other diseases. Although these approvals based solely on uncontrolled data are a relatively small proportion of the total number approvals, in absolute terms they number ∼3 per year by the EMA and 4 per year by the FDA and are worthy of further scrutiny.
The disease areas where uncontrolled studies are used for approval were primarily in oncology, with 49/74 indications (66%) being either haematological or solid tumour oncology. This also corresponds with previous work regarding drug licensing, which shows a lower barrier to oncology drug approval.9 This particularly seems to be the case with FDA reviews; of the nine applications to the EMA that were not approved, seven were in oncology, with the EMA highlighting uncertainty regarding the benefit to the risk ratio of the treatments. While the perceived unmet need in oncology has been used as an argument for the use of uncontrolled studies, we are not aware of a similar discussion in the literature regarding rare diseases. In addition, it is also possible that the FDA did not approve other treatments approved by the EMA, and this was dealt with confidential letters10 (with no press releases issued by the companies concerned). While this is unlikely, it remains an unavoidable limitation of the study.
While there is a considerable overlap in decisions, it appears that the FDA is more willing to approve products on the basis of uncontrolled trials. This difference may reflect the systems of approval or the attitudes of regulators towards risk versus unmet medical need.11 ,12 Other evidence also shows that the two systems produce decisions on different timescales, despite access to the same clinical studies. A difference in approval times has been seen in the approval of tyrosine kinase inhibitors,13 cancer drugs14 and pharmaceuticals in general15 by the two agencies. The difference may in part be explained by the FDA's extensive use of ‘accelerated approvals’ (results based on a surrogate end point, with confirmatory RCTs conducted subsequently16), compared to the lesser use of the equivalent EMA process, conditional approval.
The results regarding dates of approvals appear to show that patients in the EU must wait longer for novel treatments. The delay in submitting between agencies is understandable, given that the staff involved (eg, trial statisticians) may be required to work on submissions for both agencies. Companies appear to prioritise the FDA submission, which we have noted but not speculated on. Initiatives to harmonise the processes (eg, the Transatlantic Trade and Investment Partnership) may be helpful in reducing these delays, provided that they do not have other negative externalities.17
The issue of longer approval times for the EMA is more complex. The longer process in Europe may be due to the organisation required for making decisions for all EU member states (as well as Iceland, Liechtenstein and Norway). A further potential factor in the higher non-approval rate by the EMA (9 vs 1 applications of similar content) and the longer review period is the opportunity for discussion about the content of applications (and resolution of issues) before a dossier is submitted. Companies typically engage after each phase of clinical development with the FDA, as well as before submitting a New Drug Application.18 In Europe, companies do not necessarily interact with the EMA during the course of development but may take Scientific Advice prior to submitting; however, only 14 of the 34 products approved by both agencies had done so (Personal communication, European Medicines Agency).
Thus far, we have not discussed the appropriateness of approvals without supporting RCT results. The commonly stated factors affecting the availability of evidence are the rarity of the condition and the absence of approved treatments. The appropriateness of these arguments varies as comparative trials under rare conditions are certainly challenging; however, they do not appear to be consistent—for example canakinumab for Cryopyrin-associated periodic syndrome, with a prevalence of <1/100 000, was studied in an RCT of 31 patients. Where there is no approved standard of care in a terminal disease, in some cases (eg, alemtuzumab in relapsed and refractory chronic lymphocytic leukaemia) this justification was used for the conduct of an uncontrolled study, whereas under similar conditions (pixantrone for aggressive relapsed B-cell non-Hodgkins lymphoma) the manufacturer conducted a trial against the treatment of physicians’ choice (from six widely used off-label treatments). Even under the same conditions, there is variability in the evidence base provided by companies—Gaucher's disease is a rare lysosomal storage disorder and here taliglucerase alfa appears in our sample without controlled trial results, yet velaglucerase alfa included an active controlled trial in its development programme.
Even where placebo RCTs are clearly unviable—serious conditions without licensed treatments and a well-established outcome (poisoning is the obvious example); new treatments are often trialled against standard care or as an addition to existing therapy. Such controlled study designs have been used extensively in the evaluation of novel treatments for schizophrenia and, although not without challenges, could be used to assess how a new treatment is performed.19
While much has been written on what level of effect requires an RCT;4 in the treatments we identified, we found that the persuasiveness of evidence is highly variable. For example, imatinib in chronic myeloid leukaemia (CML) showed a response rate of 49% for major cytogenetic response and 30% for complete cytogenetic response in 532 patients, whereas omacetaxine, also in CML, showed an 18.4% major cytogenetic response rate and a 7.9% complete cytogenetic response in a trial of 76 patients. It is clear that there is a role for approval without the support of RCT results; however, what an acceptable data package should consist of does appear to be inconsistent. This is even more apparent where products must be assessed by organisations such as the National Institute for Health and Care Excellence (NICE) who has the role of estimating the difference in outcomes between the new treatment and standard of care; that is, demonstration of efficacy alone is not sufficient.20 If the studies used to gain marketing authorisation for products are also not able to provide evidence for market access (ie, to convince payers that such products represent value for money), then in some cases, the use of uncontrolled trials may be increasingly difficult to justify as they do not provide the evidence needed for the medicine to ultimately treat patients.
The research that has been conducted on treatments approved without RCT results (limited to oncology drugs approved in the USA) indicates that the availability of RCT evidence does not typically affect the likelihood of remaining on the market, with some treatments withdrawn for a lack of efficacy or safety regardless of whether randomised trial evidence was available.21 Those concerned about the lack of confirmatory evidence of efficacy will find little solace in this finding. However, the reassessment of newly licensed drugs may increase in importance with initiatives such as adaptive licensing.22 The limited information we collected (on five drugs approved on uncontrolled data by the FDA and then approved by the EMA after the availability of RCT results) suggest that current regulatory assessments may well reach sound decisions in the absence of RCT results, but this is limited by a small and selected sample. Of particular concern, however, is the extensive use of ‘historical controls’ against which to compare outcomes of trials. Previous research has highlighted that comparisons of this type are prone to exaggeration of the treatment effect, with historical controls often performing far worse than equivalent arms in clinical studies.23–25
Conclusion
Newly licensed pharmaceutical indications are frequently approved without any controlled trial results, particularly in solid and haematological malignancies. While some agents showed such high levels of effectiveness that an RCT is unlikely to have changed any decision, the level of evidence provided by companies to support marketing authorisations does appear to be inconsistent. In the more complex cases (particularly those treatments not approved by one agency), it is not always clear that the conduct of an RCT was impractical.
Although there appears to be a slight difference between approvals in the EU and the USA (with the FDA more likely to grant a positive approval in oncology), the agencies broadly reach the same decisions. However, the systematic differences in approval timing are of particular interest and concern. We therefore suggest that regulatory agencies should continue to harmonise processes where practical (to reduce delays between submissions). In parallel to this, given the frequency at which uncontrolled studies occur, their role in drug approval requires reappraisal, with formal guidance on what represents an acceptable data package; this should then be debated by regulators, companies and physicians.
References
Footnotes
Contributors AJH is a health economist specialising in the modelling and health technology assessment of pharmaceuticals, GB a Reader in Statistics and Health Economics at the University College London specialising in Bayesian biostatistics, JAB was a Professor of Biostatistics before moving to epidemiology department in Johnson & Johnson, AI is a Clinical Pharmacologist who sits on the Committee for Human Medicinal Products of the European Medicines Agency, NF is a Professor of Clinical Epidemiology & Biostatistics with over 25 years experience in clinical trials and regulatory affairs. The review of treatments was conducted by AJH with data taken from the websites of the EMA and FDA, with the article jointly written by all authors. NF is the guarantor.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests AJH works for Bresmed, an agency which undertakes contract research for the pharmaceutical industry. GB holds a research grant from Mapi, a consultancy company working in the area of health economic evaluation and has consulted with Sanofi Pasteur, GSK and Novartis on economic analysis of pharmaceutical products. JAB is a full-time employee of Johnson & Johnson and holds stock. AI is employed by the Estonian Agency of Medicines (EAM) and is a member of the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use. NF has consulted with Ipsen which has conducted single-arm trials of products for regulatory purposes, and with Sanofi Aventis, Lundbeck and Novo Nordisk on the design of randomised trials. The views expressed in this article are those of the authors and should not be interpreted as those of their employers.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data included in the article are in the public domain and freely available to others. No additional data are available.