Article Text

Abstract
Introduction The prevalence of paediatric obesity is increasing, and with it, lifestyle-related diseases in children and adolescents. High-intensity interval training (HIIT) has recently been explored as an alternate to traditional moderate-intensity continuous training (MICT) in adults with chronic disease and has been shown to induce a rapid reversal of subclinical disease markers in obese children and adolescents. The primary aim of this study is to compare the effects of HIIT with MICT on myocardial function in obese children and adolescents.
Methods and analysis Multicentre randomised controlled trial of 100 obese children and adolescents in the cities of Trondheim (Norway) and Brisbane (Australia). The trial will examine the efficacy of HIIT to improve cardiometabolic outcomes in obese children and adolescents. Participants will be randomised to (1) HIIT and nutrition advice, (2) MICT and nutrition advice or (3) nutrition advice. Participants will partake in supervised exercise training and/or nutrition sessions for 3 months. Measurements for study end points will occur at baseline, 3 months (postintervention) and 12 months (follow-up). The primary end point is myocardial function (peak systolic tissue velocity). Secondary end points include vascular function (flow-mediated dilation assessment), quantity of visceral and subcutaneous adipose tissue, myocardial structure and function, body composition, cardiorespiratory fitness, autonomic function, blood biochemistry, physical activity and nutrition. Lean, healthy children and adolescents will complete measurements for all study end points at one time point for comparative cross-sectional analyses.
Ethics and dissemination This randomised controlled trial will generate substantial information regarding the effects of exercise intensity on paediatric obesity, specifically the cardiometabolic health of this at-risk population. It is expected that communication of results will allow for the development of more effective evidence-based exercise prescription guidelines in this population while investigating the benefits of HIIT on subclinical markers of disease.
Trial registration number NCT01991106.
- Paediatric obesity
- Myocardial function
- Vascular function
- Visceral adipose tissue
- High intensity interval training
- NUTRITION & DIETETICS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- Paediatric obesity
- Myocardial function
- Vascular function
- Visceral adipose tissue
- High intensity interval training
- NUTRITION & DIETETICS
Strengths and limitations of this study
To the best of our knowledge, this multicentre trial is the first to use a combined exercise and nutrition programme to examine the effect on myocardial function in obese children and adolescents. It is also one of few trials to explore the efficacy and feasibility of high-intensity interval training in this population.
Strengths of this multicentre trial lie in the rigour of the 12-week exercise and nutrition intervention. The majority of exercise sessions will be supervised to ensure that the correct exercise intensity is achieved at all times.
Extensive resting and exercise outcome measures will be performed at both trial centres. Several measures are sensitive to small but important longitudinal changes and are novel in paediatric obesity.
Like any paediatric longitudinal trial, challenges remain around growth and maturation over the trial period, which have the potential to confound study end points. While the primary study end point is related to cardiac growth, any changes in cardiac size over the 1-year programme will be accounted for using normalisation methods.
Maturational changes will also be accounted for through Tanner stages of puberty; however, these may be subject to self-report error.
Introduction
Paediatric obesity rates have increased over the last two decades and is now prevalent in 3–15% of the paediatric population according to the International Obesity Task Force definition of obesity.1 More than 50% of obese children will become obese adults2 with a significantly increased risk of developing non-communicable diseases, including cardiovascular diseases, cancer and type 2 diabetes mellitus (T2DM).3 In 2012, these diseases accounted for 63% of deaths worldwide3 and 77% of disease burden in Europe.4 Indeed, a childhood and adolescent body mass index (BMI) above the 95th centile is a strong predictor of adult mortality rates.5 Furthermore, children and adolescents who had a baseline BMI between the 85th and 95th centiles, defined as overweight, had a 30% increase in all-cause mortality. This increased risk of death was independent of their adult BMI.5
Obese children and adolescents may show abnormal myocardial function when assessed through resting and stress echocardiography.6–10 Echocardiographic techniques such as tissue Doppler imaging are able to detect subclinical heart disease.11 Previous investigations have shown significantly reduced tissue Doppler velocities in obese youth compared with lean, healthy age-matched control participants.12 In particular, peak systolic tissue Doppler velocity (S′) closely reflects left ventricular contractility,13 which can be improved with short-term exercise training.14
Increased BMI or overweight in early life (1–9 years) is associated with coronary artery disease15 and it is acknowledged that atherosclerotic processes begin in childhood.16 Impaired vascular function determined by flow-mediated dilation (FMD) of the brachial artery has been observed in a number of obese paediatric studies.17–22 Visceral adipose tissue is increased in obesity which results in a greater release of bioactive mediators.23 These influence the function of adipose tissue and contribute to chronic disease, having a substantial impact on insulin sensitivity, inflammation and subsequent risk for dyslipidaemia, T2DM and atherosclerosis.24
Current paediatric guidelines for treating paediatric obesity recommend lifestyle modification to encourage family-based behaviour change leading to a reduction in energy intake and increase in physical activity.25 ,26 The current treatment of paediatric obesity appears to have a low success rate, most likely due to the heterogeneous causes of obesity. Several recent meta-analyses and reviews have demonstrated that the effectiveness of paediatric obesity treatment is limited.27–29 The response to treatment differs substantially between clinical centres and treatment success appears to be age related. A large European registry study showed significant treatment effects following a variety of lifestyle interventions, including exercise programmes, nutrition education, psychological intervention and parental education, in <10% of participants over 2 years of follow-up.30 Furthermore, the treatment was least effective in participants older than 12 years. However, the low success rate was in part accounted for by a high dropout rate. Two recent reviews suggest that lifestyle and exercise interventions in obese children and adolescents can lead to improvements in anthropometric and cardiometabolic outcomes. These reviews are not inclusive of several important outcomes such as myocardial and vascular function, visceral adipose tissue or cardiorespiratory fitness.27 ,31 Myocardial and vascular function outcomes have prognostic significance,32 ,33 are able to identify subclinical disease7 ,34 ,35 and may be improved with exercise training.12 ,36 There is growing evidence that paediatric obesity is associated with subclinical structural and functional cardiovascular alterations.12 ,35 Myocardial dysfunction is more easily unmasked during stress as it precedes resting abnormalities.12 ,37 Furthermore, low cardiorespiratory fitness has shown independent associations with all-cause mortality38 and improvements in cardiorespiratory fitness may attenuate risk of metabolic disease39 independent of visceral adipose tissue.40 There is minimal evidence regarding the effect of exercise intensity on these novel markers in paediatric obesity.
Current worldwide data show that <50% of children and adolescents accumulate the minimum recommended 60 min of moderate-to-vigorous-intensity physical activity every day.41–46 Moreover, obese children spend approximately 100 min a day being less physically active than healthy weight or overweight children.47 The promotion of high-intensity exercise in this obese paediatric group may be an alternative to improve cardiometabolic outcomes. High-intensity interval training (HIIT) has recently been explored as an alternate exercise to moderate-intensity continuous training (MICT) in healthy adults as well as those with chronic disease. HIIT involves a short bout of exercise at a high intensity, interspersed by recovery periods in preparation for the next bout. HIIT has resulted in improved health markers in adults with cardiometabolic disease48 while demonstrating time efficiency.49 Four studies to date have examined the physiological effects of HIIT in obese children and adolescents and reported positive findings.12 ,50–52 Children and adolescents expressed increased enjoyment during HIIT49 and the stop–start nature of HIIT may reflect play-based activities traditionally observed during childhood. This is particularly important as enjoyment is a strong determinant of exercise adherence.53 ,54 In fact, a ‘lack of enjoyment’ is frequently reported as a barrier by obese children.55 While the chosen HIIT protocol (40 min; 10 min warm-up, 4×4 min intervals interspersed by 3 min active recovery, 5 min cool down) has a similar time commitment to the MICT protocol (44 min), this is a result of equalising energy expenditure between training types.56 We have previously shown that this particular HIIT protocol in obese adolescents can almost normalise cardiac function to that observed in lean counterparts, and high compliance to the HIIT protocol was noted.12 However, the pilot trial had a small sample size and did not include comparative treatments.12 The 4×4 HIIT protocol has shown great efficacy in clinical adult populations, including patients with heart failure, coronary artery disease, hypertension, obesity and the metabolic syndrome.48 Weston et al48 recommend the use of this protocol due to greater time efficiency , that is, increases in cardiorespiratory fitness following a HIIT programme were nearly double the increases seen after a MICT programme (19.4% vs 10.3%). In the studies reviewed, Weston et al48 found that HIIT was able to elicit many superior benefits to MICT, albeit in only a slightly shorter time period. This randomised controlled trial aims to examine the physiological efficacy of HIIT compared with MICT in a multicentre randomised controlled trial, thereby improving the current literature and informing the treatment options for paediatric obesity.
Objectives
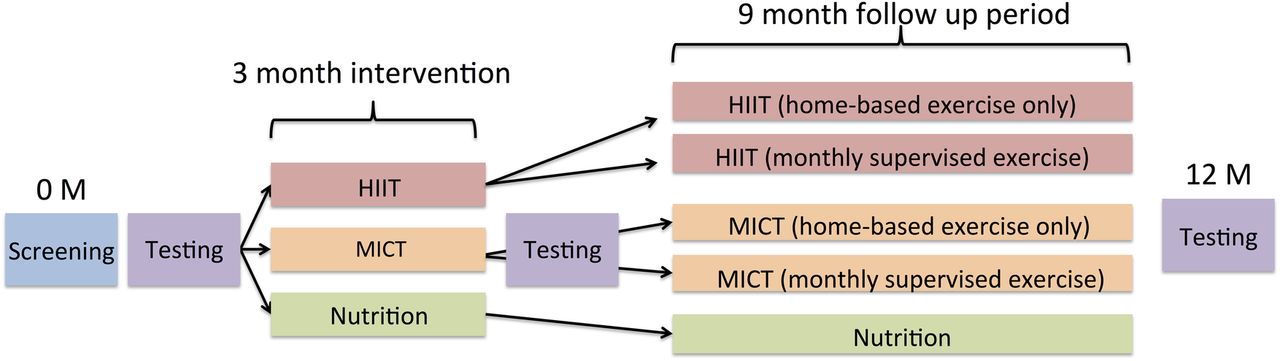
The primary aim of this randomised controlled trial is to compare the effectiveness of three 12-month interventions: HIIT and nutrition advice, MICT and nutrition advice or nutrition advice alone on myocardial function in obese children and adolescents. It is hypothesised that HIIT will be superior in improving myocardial function compared with MICT. Secondary outcome measures will include vascular function, body composition (total, visceral and subcutaneous adipose tissue depots and lean muscle mass), cardiac structure, cardiorespiratory fitness, autonomic function, serum fasting lipids and insulin sensitivity, biomarkers of inflammation, satiety and oxidative stress markers, physical activity levels and nutrition. Phase I of the trial will examine the effectiveness of an intensive 3-month period on the stated outcomes with assessments at baseline and after 3 months of supervised training. Phase II of the trial aims to determine the amount of supervision required to maintain exercise habits over a 9-month home-based training period. For this phase, participants will be rerandomised to (1) monthly supervised exercise or (2) home-based exercise only. Final assessments will be completed at 12 months. Lastly, the trial aims to determine whether any of the intervention arms are able to improve and normalise outcomes comparable with those found in healthy weight children and adolescents. It is for this comparative purpose that an age-matched, lean and healthy control group of children and adolescents will be assessed at a single time point.
Methods and analysis
Study setting
This is a multicentre randomised controlled intervention trial. The study centre is located at the Norwegian University of Science and Technology (NTNU), Trondheim, Norway. The other centre is at the University of Queensland (UQ), Brisbane, Australia. Testing and training will take place in the university research laboratories at these institutions and hospital outpatient settings (St. Olav's Hospital in Trondheim and the Wesley Hospital in Brisbane).
Participants and eligibility criteria
The study cohort will include 100 obese and 100 lean, healthy control children or adolescents aged between 7 and 16 years old. Obesity in this population will be defined as a BMI equal to or greater than the 95th centile (age and sex specific).57 Study exclusion criteria include hypertension (defined as blood pressure above the 95th centile for systolic or diastolic values), any history or evidence of heart disease and/or an abnormal resting or stress echocardiography which indicates it would be unsafe to participate, any chronic disease, for example, chronic asthma, kidney disease, diabetes, current smoking habits or an orthopaedic/neurological disorder that may limit ability to exercise, diagnosed attention deficit hypersensitivity disorder and use of steroid medications. Conditions not specifically aforementioned may serve as criteria for exclusion at discretion of the clinical site. Furthermore if medical conditions become apparent in participants during assessments or exercise training, medical advice will be sought and the intervention may be discontinued in the individual. Lean healthy control participants must have an age and sex-specific BMI in the healthy range (5th–85th centile) to be included.57 Study exclusion criteria are identical in lean healthy control participants. The lean healthy control participants will not partake in an intervention but will be assessed for cross-sectional comparative analyses.
Interventions
A randomised block design will be used. Obese participants will be randomised to one of three groups and will be stratified according to age and sex. Lean participants will be stratified to the same groups but no intervention will be administered (figure 1).

Intervention groups/stratification.
Exercise protocol
The exercise intervention involves a combination of supervised and unsupervised exercise training sessions. Participants will attend at least two, up to three, supervised training sessions each week for 12 weeks. If participants choose to attend two supervised sessions, they will be required to complete a third unsupervised exercise session at home. Following the 3-month supervised period, participants in each of the two exercise groups will be randomised to ‘monthly supervised exercise’ or ‘home-based exercise only’ from 3 to 12 months. During this time, participants in the HIIT and MICT groups will be asked to complete three unsupervised training sessions each week for 9 months and the ‘monthly supervised exercise’ group will be asked to attend once monthly supervised training sessions at the clinical site.
Supervised exercise training (HIIT and MICT) will consist of walking or running on a treadmill or cycling on a stationary bike based on participant preference. During the unsupervised exercise session, the mode can vary. Necessary adjustments to speed/grade and resistance will be made over the course of the intervention to ensure that target heart rate zones are achieved at all times. Heart rate, rating of perceived exertion (RPE) (Pictorial Children's Effort Rating Table—PCERT) and exercise mode will be recorded in a training booklet during supervised and unsupervised exercise sessions. In the event of limited access to a heart rate monitor during unsupervised sessions, participants will be asked to replicate the supervised session as closely as possible (ie, identical treadmill speed/grade for intervals and active recovery periods) and to aim for similar RPE recordings as in the supervised sessions. Participants who wish to complete an unsupervised session each week will be provided with a separate booklet, which will be kept alongside the clinic version. Each participant will also receive a training booklet for the follow-up period allowing them to record details of each session completed.
High-intensity interval training
Participants randomised to HIIT will perform a 10 min warm-up at 60–70% of maximal heart rate (HRmax). Following this, they will walk, run or cycle at 85–95% of their HRmax for four, 4 min intervals, with 3 min of active recovery (50–70% of HRmax) between the intervals. Participants will perform a 5 min cool down period at the end amounting to a total exercise time of 40 min.
Moderate-intensity continuous training
Participants randomised to the MICT group will walk, run or cycle continuously at 60–70% HRmax for 44 min to approximate the average energy expended by the HIIT group as previously calculated by our research group.56
Nutrition advice
HIIT, MICT and nutrition groups will receive 8 to 10, 20 min individual nutrition sessions with a dietitian over the 12-month period. Content of the sessions will include healthy food choices, portion sizes and regular meal times. The nutritional advice given will reflect current Norwegian and Australian eating guidelines and will be location specific.58 ,59 The nutrition group will not be provided with any prescribed supervised exercise.
Outcomes
Measurements will take place at baseline, 3 months (end of the supervised exercise intervention period) and at 12-month follow-up.
Participant preparation guidelines
Participants will be informed of preparation requirements, which will be checked prior to the assessments.
Stress echocardiography and cardiorespiratory fitness testing
Assessment of myocardial function and structure and maximal oxygen uptake (VO2max) will require participants to refrain from eating heavy meals up to 2 h before the testing session and avoid any foods containing caffeine for this time period.
Vascular function, body composition and blood collection
Assessment of vascular function and body composition (BodPod), and blood collection will be completed in a fasted state (8–12 h) and participants will be instructed to avoid caffeine, vitamin C, alcohol, drugs, stimulants and medications for this time period. Additionally, participants must refrain from intense exercise for 48 h prior to testing. To avoid dehydration, participants will be instructed to drink at least 0.5 L of water before attending the assessment.
Myocardial function (rest and stress echocardiography)
Primary outcome measure: S′ at rest (left ventricle).
Secondary outcome measures: S′ (during exercise, both ventricles), S′ (rest, right ventricle), peak diastolic tissue velocities (both ventricles), tricuspid annular plane systolic excursion (TAPSE), global strain and strain rate, aortic flow and cardiac dimensions.
A full resting echocardiogram will be performed with a Vivid 7/E9 ultrasound machine (GE Vingmed Ultrasound, Horten, Norway) using a phased array transducer (GE M3S). Three cine loops from the three standard apical planes (four chamber, two chamber and long axis) and the right ventricle will be recorded in grey-scale harmonic mode and tissue Doppler mode simultaneously. Left ventricular (LV) standard Doppler echocardiographic indices will be measured and body surface area (m2) will be used to normalise cardiac dimensions for differences in body size. Mitral annulus excursion, pulsed wave tissue Doppler velocities, peak systolic (S′), peak early diastolic (e′) and peak late diastolic (a′) will be measured at the atrioventricular (AV) plane level in four-chamber and two-chamber view and a mean of the four points will be used. Standard Doppler echocardiographic indices of right ventricular function will be measured as well as TAPSE, S′, e′, a′. Deformation (strain) and deformation rate (strain rate) will also be analysed by speckle tracking (2D strain) and tissue Doppler imaging.
Following the resting echocardiogram, individuals will exercise on a stationary cycle ergometer in an upright position. The exercise protocol will start at an intensity of 25 W with 25 W increments every 3 min until participants have attained their HRmax or are no longer able to maintain a constant cadence. Recordings will be made at baseline and peak assessing apical four chamber and two chamber in B-mode and tissue Doppler as well as mitral and aortic flow. A three-lead ECG, blood pressure and ratings of perceived exertion (PCERT) will be monitored and recorded at the end of each stage.
EchoPAC (GE Vingmed Ultrasound AS, Horten, Norway) will be used for all echocardiographic analysis by an investigator blinded to the group allocation of the subjects.
Visceral, subcutaneous and total abdominal adipose tissue (MRI)
At the Brisbane site, visceral, subcutaneous and total abdominal adipose tissue will be measured using a 1.5 T MRI system (Siemens Symphony Sonata, Siemens, Erlangen, Germany) equipped with a six-channel body matrix coil and a six-channel spine coil. Subjects will be positioned supine inside the magnet and transversal images will be acquired using true fast imaging steady state precision (TRUFISP) technique with breath hold (repetition time=3.76 ms; echo time=1.88 ms; flip angle 75°; matrix=220×256; rectangular field of view (FOV)=400 mm×400 mm; slice thickness=8 mm; 14 slices; acquisition time=12 s). We will acquire 14 axial slices, 8 mm thick centred over the umbilicus during breath hold using a Dixon technique.
At the Trondheim site, visceral, subcutaneous and total abdominal adipose tissue will be measured using a 3 T MRI system (Siemens Skyra; Siemens, Munich, Germany) equipped with an 18-channel body coil and a 32-channel spine coil. Participants will be positioned supine inside the magnet and transversal images will be acquired using T1-weighted Dixon vibe sequences with breath hold (repetition time=4.04 ms; echo time=1.3 ms and 2.50 ms; flip angle=9°; matrix=320×256; rectangular FOV=380 mm×309; slice thickness=3 mm; 52 slices; number of averages=1; band width=1120 Hz/pixel; generalised autocalibrating partially parallel acquisition (GRAPPA) parallel imaging acceleration factor 2, acquisition time=17 s). If the given FOV is too small to cover the whole subject, the FOV is increased sufficiently before scanning. The Dixon sequences will be acquired twice to assure a set of successful images.
The MRI scans will be exported and anonymised. Images will be converted to NIfTI format and analysed using in-house software developed in MATLAB (TheMathWorks, Inc, Natick, Massachusetts, USA). Five consecutive slices at and above the L4/L5 vertebral disc, with localising images used to confirm the level, will be selected for quantification of visceral and subcutaneous fat in each participant. Two regions of interest (ROIs) will be manually drawn on the images by a trained radiologist. One ROI will delineate the subcutaneous compartment and the second ROI will delineate the intra-abdominal and retroperitoneal areas together to quantify visceral adipose tissue. Visceral and subcutaneous adipose tissue will be summed to quantify total adipose tissue. The area of the fat inside each ROI will be calculated by counting the number of pixels with intensities above a selected threshold and multiplied by the pixel area. Slices will be manually adjusted for threshold intensity in order to compensate for lack of uniformity between slices. Values calculated from the five slices will be averaged to provide a mean area (cm2).
Vascular function (FMD)
Endothelial function of the brachial artery will be measured via FMD using high-resolution vascular ultrasound (12–14 MHz ultrasound Doppler probe, Vivid 7 system/Vivid I system; GE Vingmed Ultrasound AS, Horten, Norway) according to current guidelines.60 Participants will lie supine for 10 min in a quiet, dark environment prior to the start of the procedure. A transducer will be placed against the brachial artery and following image optimisation, baseline images will be recorded for at least 30 s in live duplex mode (simultaneous B-mode diameter and pulsed wave Doppler velocity signals). A blood pressure cuff will then be placed distal to the region of interest and inflated for 5 min at 200 mm Hg. This will reduce blood flow to the hand. Upon cuff release, 10 beat cine loops will be recorded in duplex mode (B-mode diameter and pulsed wave Doppler velocity signals) at 10, 30, 60, 90, 120, 150 and 180 s (UQ), or continuously for 3 min (NTNU) to measure the change in the diameter of the artery following increased blood flow and shear stress to the vascular wall. Data will be assessed by custom-made automated edge-detection software, which is independent of investigator bias. Brachial artery diameter and shear rate will be quantified from the ultrasound studies to assess FMD.
Cardiorespiratory fitness
VO2max will be measured during uphill treadmill walking or running using expired air gas analysis (Metamax3B, Cortex Biophysik, GmbH, Leipzig, Germany or Jaeger Oxycon Pro, CareFusion, Hoechberg, Germany). An incremental, ramp protocol will be used for all participants. A 4 min warm-up at 4 km/h and 0% grade will precede the test; however, the speed can be modified to match the preferred walking speed of the participant. A walking or running protocol is available depending on participant preference and fitness.
The walking protocol consists of 1 min stages where speed is set at preferred walking speed and gradient is increased by 2% each minute. If the participant reaches a steep gradient (this will be adjusted by researchers depending on age and height of participant: <12 years approximately 12%, ≥12 years approximately 16%), speed will be increased by 1 km/h each minute thereafter.
The running protocol consists of 1 min stages where gradient is set at approximately 10% (this will be adjusted by investigators depending on age and height of participant) and speed is increased by 1 km/h each minute. If the participant reaches a speed that can no longer be safely increased, gradient will be increased by 1% each minute thereafter.
A levelling off of oxygen uptake (VO2) despite increased workload and respiratory exchange ratio (RER) ≥1.05 will be used as criteria for VO2max.
A levelling off (plateau) in VO2 will be defined using 30 s epochs. If the VO2 increase is  with an increase in workload, then a plateau is assumed. If the participant has reached a plateau and the RER criterion has been satisfied then the two highest consecutive 30 s values will be averaged to obtain the VO2max value. If the participant has not reached a plateau, VO2peak is determined by using the average of the two highest values attained. It is highly likely that most participants will not reach a VO2max, and therefore group values will be reported as VO2peak.
with an increase in workload, then a plateau is assumed. If the participant has reached a plateau and the RER criterion has been satisfied then the two highest consecutive 30 s values will be averaged to obtain the VO2max value. If the participant has not reached a plateau, VO2peak is determined by using the average of the two highest values attained. It is highly likely that most participants will not reach a VO2max, and therefore group values will be reported as VO2peak.
Heart rate will be measured continuously during the test (Polar, Polar Electro, Kempele, Finland) to define HRmax.
Heart rate variability
Participants will lie supine for 10 min in a quiet, dark environment prior to commencement of the procedure. Participants will be asked to lie as still as possible for 5 min while an ECG trace is monitored and recorded for calculation of heart rate variability. RR intervals obtained from the ECG will be processed using Kubios HRV (University of Eastern Finland, Finland).
Body composition
At the Brisbane site, dual energy X-ray absorptiometry (DXA) will be used to determine body composition (adipose tissue and lean muscle mass). This will require the participant to lie motionless while an X-ray of their entire body is taken using the DXA scanner (Hologic, QDR Series, Massachusetts, USA). The duration of a whole body scan is 7 min.
At the Trondheim site, a BodPod (COSMED, Rome, Italy) will be used to determine body composition (adipose tissue and lean muscle mass). Participants are required to be fasted (8 h) and will be tested in minimal clothing (underwear only). The procedure takes 15 min in total with 5 min spent in the BodPod.
Blood biochemistry
Venous blood samples will be collected from a superficial antecubital vein according to standard phlebotomy procedures. Samples will be collected into three vacutainers containing EDTA, fluoride oxalate and clot activators. Vacutainers will be stored on ice or left to clot at room temperature for at least 30 min (serum samples). Two aliquots of whole blood will be pipetted out and following this, samples will be centrifuged at 2500 rpm, 4°C for 10 min. Plasma/serum will be aliquoted and stored at −80°C for later analysis.
Samples will be analysed for lipids (total cholesterol, low-density lipoprotein (LDL), high-density lipoprotein (HDL), triglycerides), glucose and insulin (to establish insulin resistance and β cell function using Homeostatic Model Assessment of Insulin Resistance (HOMA-IR)) and C reactive protein (CRP) using spectrophotometry (Cobas Mira, Roche Diagnostics, Australia). Insulin resistance and β cell function will be calculated using the HOMA model (based on fasting glucose and insulin concentrations), where  and
and  . Satiety hormones (ghrelin, leptin, peptide YY, obestatin), inflammatory markers (tumour necrosis factor α (TNFα), interleukin 6 (IL-6), IL-10, plasminogen activator inhibitor 1 (PAI-1)), adiponectin and soluble intercellular adhesion molecule 1 (si-CAM-1) will be measured using specific ELISA kits (R&D systems, Inc, Minneapolis, Minnesota, USA). Total nitrite concentration will be quantified using a commercially available assay for nitric oxide (NO2−) detection (R&D systems, Inc, Minneapolis, Minnesota, USA). Oxidative stress and antioxidant status will be measured using the following methodologies. Total F2-isoprostanes will be extracted and analysed using a method developed by the Brisbane laboratory.61 The laboratory coefficient of variation for this assay is 4.5%. Protein carbonyls will be analysed using adapted methodology from Levine et al.62 The laboratory coefficient of variation for this assay is 11.9%. Plasma glutathione peroxidase (GPx) activity will be measured spectrophotometrically (Cobas Mira, Roche Diagnostics, Switzerland) via the oxidation of NADPH to NADP by modifying methods.63 ,64 The laboratory coefficient of variation for this assay is 2.4%.
. Satiety hormones (ghrelin, leptin, peptide YY, obestatin), inflammatory markers (tumour necrosis factor α (TNFα), interleukin 6 (IL-6), IL-10, plasminogen activator inhibitor 1 (PAI-1)), adiponectin and soluble intercellular adhesion molecule 1 (si-CAM-1) will be measured using specific ELISA kits (R&D systems, Inc, Minneapolis, Minnesota, USA). Total nitrite concentration will be quantified using a commercially available assay for nitric oxide (NO2−) detection (R&D systems, Inc, Minneapolis, Minnesota, USA). Oxidative stress and antioxidant status will be measured using the following methodologies. Total F2-isoprostanes will be extracted and analysed using a method developed by the Brisbane laboratory.61 The laboratory coefficient of variation for this assay is 4.5%. Protein carbonyls will be analysed using adapted methodology from Levine et al.62 The laboratory coefficient of variation for this assay is 11.9%. Plasma glutathione peroxidase (GPx) activity will be measured spectrophotometrically (Cobas Mira, Roche Diagnostics, Switzerland) via the oxidation of NADPH to NADP by modifying methods.63 ,64 The laboratory coefficient of variation for this assay is 2.4%.
Physical activity and nutrition measurements
Accelerometry will be used to measure physical activity at baseline and at the 3-month assessment. Participants will be asked to wear an accelerometer for 7 days (Brisbane: ActiGraph, Florida, USA and Trondheim: SenseWear, BodyMedia, Inc, Pittsburgh, Pennsylvania, USA). To be included in the analysis, participants will be required to have a minimum of 4 valid days, 1 of which must be a weekend day. Participants in Brisbane will be asked to wear the ActiGraph monitor during waking hours, except for when sleeping or during water-based activities. Participants will also be asked to keep a brief log to record wake/sleep times and any time the monitor was removed for >10 min (eg, sleep, shower, etc). The ActiGraph accelerometer will be initialised to sample at 30 Hz and data will be aggregated in 15 s epochs. To be considered a valid day, there must be a minimum of 10 h of wear time and non-wear time criteria will be 60 min or more of consecutive zeros. Accelerometer cut points previously validated in a paediatric population65 will be used to determine average time per day spent in light physical activity, moderate physical activity and vigorous physical activity. ActiGraph data will be analysed using ActiLife software (V.6). Participants in Trondheim will be asked to wear the SenseWear armband for 24 h each day. The device will be removed during water-based activities. To be considered a valid day, at least 85% of a 24 h day must be recorded and time spent in light physical activity (1.6–2.9 metabolic equivalents (METS)), moderate physical activity (3–5.9 METS) and vigorous physical activity (>6 METS) will be determined. SenseWear data will be analysed using BodyMedia (V.8.1, Pittsburgh, Pennsylvania, USA).
Participants will also complete a physical activity questionnaire designed by the Norwegian Directorate of Health with content focusing on total physical activity, physical activity at school and home and weekday/weekend screen time.66
Food record booklets will be given to participants and a 3–4-day record will be requested (must include 1 weekend day). Food records will be analysed using either FoodWorks (Xyris Software, Australia) for the Australian cohort or using the food database KBS AE-07 and KBS software system (KBS, V.4.9, 2008, Department of Nutrition, University of Oslo, Norway) for participants in Norway.
Other measures
Height, weight, waist circumference (WC) and hip circumference (HC) and blood pressure will be measured using standard approaches.67
Participant timeline
Figure 2 illustrates the schedule for measurement of outcomes and the intervention.

{kind=link}
{kind=link}
A scheme illustrating a time schedule for enrolment, intervention and assessment of obese participants.
Sample size
The sample size was calculated for a one-way analysis of variance (ANOVA) comparing the mean change in resting S′ between the three groups from preintervention to postintervention. Although the data will be analysed using a linear mixed model (LMM), a simplified calculation of sample size for a one-way ANOVA provides a conservative estimate of the sample size required for the LMM. Simulations run by our research group support this assumption. For calculation of sample size, the clinically meaningful difference in means was set to 1 cm/s, using the values 9.5, 10 and 10.5 cm/s for the nutrition, MICT and HIIT groups, respectively. The SD, assumed to be equal for all groups, was set to SD=0.9 cm/s.12 To obtain a power of 0.80 for the overall test of differences in means, using a significance level of 0.05, 17 individuals are required in each group. A further 40 individuals are required to account for the four stratification groups (figure 1) included in all statistical analyses. To account for 15% dropout, a total of 105 individuals are required to enter the intervention. In order for the lean control group to be closely matched in age, sex and sample size, 105 lean participants are required for assessment at a single time point.
Recruitment
A variety of recruitment strategies will be employed at each site to achieve adequate participant enrolment and reach target sample size. At the Trondheim site, a regular advertisement will be placed in the local newspaper. In addition, advertisements will be published on the university and hospital websites and flyers will be distributed at strategic locations. Every 6 months, local health nurses and medical centres will be informed about the study. A website will be set up with a linked preliminary screening tool and programme contact form. A video blog about childhood obesity research will be used for media outlets and publicity through newspapers and television. Social media such as Facebook will also be used. Similar strategies will be used at the Brisbane site. An advertisement for the study will be placed in the university staff newsletter and flyers will be placed around the campus. Schools in a 30 km radius will be emailed with a newsletter advertisement. Health and fitness centres and medical centres within a 15 km radius will be asked to advertise the programme on a noticeboard. A website will be set up with a linked preliminary screening tool and programme contact form. Google AdWords will be employed as a continuous recruitment pathway strategy with approximately 10–15 clicks resulting in website views expected each day. Local dietitians and paediatricians will also be contacted for referrals into the programme. Finally, media outlets will be contacted for newspaper and television coverage.
Assignment of interventions
Allocation sequence generation
Computer-generated random numbers will be used for allocation sequence generation with stratification completed according to age and sex (see figure 1). The four stratification groups will be:
Females 7–10 years
Males 7–12 years
Females 10–16 years
Males 12–16 years
Allocation concealment will be implemented using central randomisation through a web-based program that will generate the allocation sequence. A study investigator at each centre will enter the details of eligible obese participants and the web-based program will provide the group allocation for the intervention. The investigator will then inform the parent or guardian. Lean and healthy control participants will also be registered and stratified using the web-based program.
Blinding
As this is an exercise intervention, trial participants cannot be blinded to group assignment. However, outcome assessors and data analysts will be blinded to intervention assignment. Outcome assessors of the primary outcome are independent of the clinical centre and will not interact with participants outside of the assessments. Participants will be asked not to divulge their group allocation during testing visits. All data is stored using participant ID number only and data analysis that is subject to investigator bias will occur without knowledge of intervention assignment.
Data management and analysis
Data management
Double data entry will be administered at each site to ensure data quality. Data will be stored in a reidentifiable format (participant numbers only). A password-protected sheet will enable the participants' numbers be linked to names when required.
During and after the research project, data on paper will be kept in a locked filing cabinet. Electronic data will be stored on password-protected computers or external hard drives with access granted only to members of the research team. Tissue samples will be identified by participant number only and will be stored in a secure locked area.
Information collected will be disposed of 10 years after study completion. Paper documents will be shredded and disposed of, while electronic information will be erased.
Statistical methods
An intention to treat analysis will be used. A per protocol analysis will also be conducted where completion of 80% of the 3-month intervention is required. Data will be analysed with SPSS Statistics (IBM, New York, USA), Stata (Texas, USA) and R (R Core Team, Vienna, Austria). Normality of data will be checked using one or more normality tests. Descriptive statistics will be computed for variables of interest and continuous data will be reported as means and SDs if data is normally distributed. Non-continuous and non-normally distributed data will be reported as frequencies, medians and IQRs.
Statistical analysis comparing between group differences (three groups) following the intervention will be conducted using an LMM. This technique calculates between-group and within-in group differences (from baseline to postintervention) within the same model. An LMM is also able to adjust for stratification variables (age, sex) and for a centre effect if present. Adjustment for a centre effect is particularly important, as there are minor discrepancies in the intervention and outcome methodologies, as well as a large climatic difference due to varying geographical locations of the clinical centres. In order to examine the effect of supervision during the 9-month follow-up period (from 3 to 12 months), an LMM will be used again with supervision included as an additional explanatory variable in the model.
To compare between-group differences in binary categorical data following the 3-month intervention, a generalised LMM (GLMM), which accounts for time correlations, will be used.
Monitoring
Harms
If a participant develops a medical or surgical illness during the study, the Data Monitoring Committee in cooperation with the participant's general practitioner will ascertain continuing or resuming participation in the intervention. In the event of a medical emergency occurring at the clinical sites, the study staff will undertake, under direction of the principal investigator or designated staff, all necessary supportive medical care.
All adverse events (AEs) will be reported between the first trial-related study procedure and the last during study intervention. Medical events that occur between the signing of the informed consent form and the final study-related procedure will be documented in the medical history.
Participants should voluntarily report any AEs or in response to general, non-directed questioning (eg, ‘How has your health been since the last visit?’). For each AE volunteered by the participant, the investigator will obtain all the information required to complete the AE documentation. All AEs regardless of seriousness, severity or presumed relationship to the study will be recorded using medical terminology in the source document and on the case report form. Safety-related events will be reported in a timely fashion as required by the Data Monitoring Committee and the Ethics Committees responsible for the study.
Ethics and dissemination
Consent and assent
Participants and their parents will provide informed consent after the principal investigator has briefed them on the study and answered questions. Two separate informed consent sheets will be signed with content adjusted to suit a paediatric population (participant and parent/guardian signs one while the other is signed by the participant only).
Confidentiality
Information collected directly from participants will be in a reidentifiable form and any information collected for, used in or generated by this project will not be used for any other purpose. The site principal investigator and associated research personnel such as the study dietitian will have access to information.
Dissemination policy
Results of this clinical trial aim to be disseminated through peer-reviewed journal articles, conference abstracts and presentations, as well as media publications. It is hoped that results of the clinical trial will inform future paediatric obesity management strategies.
Patient and public involvement
Patient and public involvement (PPI) strategies were implemented following the conclusion of preceding pilot trial. Several organisations were involved in the conception of protocol design in Brisbane (Child Obesity Program at the Mater Children's Hospital/Lady Cilento Children's Hospital and The Children's Nutrition Research) and Norway (Centre for Obese Adults and Children at St. Olav's Hospital, the Cardiac Exercise Research Group at NTNU, the Norwegian Physiotherapist Association, as well as local schools and physical education (PE) teachers in Trondheim). Six clinicians were also actively involved during the protocol development phase, including cardiologists, paediatric endocrinologists and dietitians. The conclusion of the pilot trial provided an opportunity to receive feedback from the patient population in question. While a proportion of obese adolescents disliked HIIT until they improved their cardiorespiratory fitness, the majority reported enjoyment, which resulted in high training attendance and compliance. Freedom with exercise modality also increased HIIT feasibility in this group. A similar protocol was therefore implemented in the current intervention.
During the ongoing clinical trial, feedback from participants and families has been welcomed and resulted in minor protocol adjustments. Notable adjustments include the structure of assessments and frequency of supervised exercise sessions offered in order to aid adherence.
Discussion
Reduced physical activity and poor nutrition are the main causes of obesity in children and adolescents. A large proportion of the paediatric population does not meet exercise guidelines, and energy intake in obese children is often larger than in healthy weight children.68 ,69 Consequently, paediatric obesity has increased steadily in several countries over the last 30 years.70–72 Current worldwide data suggest that only 5–50% of children and adolescents are meeting current exercise guidelines.41–46 ,73 Furthermore, obese children are less physically active compared with healthy weight children74 ,75 and spend more time in sedentary activities.76 ,77 Low levels of leisure time physical activity are associated with an increased risk of cardiovascular and metabolic diseases.78 Therefore, encouraging all children to increase their levels of physical activity and reduce their sitting time could help to avoid excess weight gain and associated health risks.79 We therefore are striving to investigate a time-efficient form of exercise, which could potentially induce physiological changes and reduce cardiovascular risk factors in obese children and adolescents. The importance of nutrition in this population stipulated that the exercise interventions were combined with nutrition advice and compared with a nutrition advice only group as well.
This clinical trial involves the largest known cohort of obese children and adolescents in a HIIT intervention to date. Valuable information about the effects of exercise intensity on cardiac and vascular structure and function, body composition, cardiorespiratory fitness, biochemistry markers, physical activity, nutrition and quality of life in obese children and adolescents will be gathered. Importantly, this trial will contribute to the currently small, but important, body of evidence exploring cardiac function during exercise. While resting function may remain normal in paediatric obesity, stress echocardiography may unmask subclinical cardiac disease35 and lead to better patient management and outcomes.
Dissemination of the knowledge gained from this trial is expected to inform physical activity and exercise guidelines for this specific population in an attempt to dampen the impact that obesity has on physiological systems as well as reduce risk factors for future development of comorbidities such as T2DM and heart disease.
References
Footnotes
Contributors CBI and AET conceptualised the trial and drafted the original study methods. KAD, JSC and CBI refined study methods and will be the project managers of this multicentre research trial. CBI and PAC expertise lie in assessment of myocardial function and structure, while DJG and MSH specifically assisted with non-invasive methodologies for assessing vascular health. MH and EMH designed the protocol for assessment of visceral and subcutaneous fat, while SEK, MH and EH developed analysis methodologies for this outcome. PSWD and SMH-S expertise lie in paediatric nutrition, and therefore contributed to design of the nutrition intervention. GML has extensive experience in paediatric endocrinology and paediatric obesity lifestyle interventions. GML assisted with general study design and identifying blood biochemistry variables of interest. SRG assisted with selecting an activity monitor device and with study design to collect and analyse physical activity levels. AET and KAD designed the methods for the maximal oxygen consumption test. TBR specifically contributed to biostatistics of the trial, including sample size and power calculations and statistical analysis methods.
Funding This work was supported by St Olav's Hospital, the Norwegian University of Science and Technology (grant number #9527) and the Wesley and St Andrew's Research Institute (grant number #2014-01). SRG is supported by an Australian National Health and Medical Research Council Program Grant (#569940) at the University of Queensland.
Competing interests JSC reports grants from Coca Cola, personal fees from Tolmar Pharmaceuticals, personal fees from Novo Nordisk Pharmaceuticals, outside the submitted work.
Ethics approval At NTNU, Norway, the Regional Committee for Medical and Health Research Ethics has approved this project (reference number 2009/1313-4). At UQ, Australia, the University of Queensland Human Research Ethics Committee (reference number 2013000539), the Mater Hospital Human Research Ethics Committee (reference number HREC/13/MHS/119/AM01) and Uniting Care Health Human Research Ethics Committee (reference number 1324) have approved this project.
Provenance and peer review Not commissioned; externally peer reviewed.