Article Text

Abstract
Objectives In this cross-sectional epidemiological study we aimed to identify molecular profiles for Trichomonas vaginalis and to determine how these molecular profiles were related to patient demographic and clinical characteristics.
Setting Molecular typing methods previously identified two genetically distinct subpopulations for T. vaginalis; however, few molecular epidemiological studies have been performed. We now increased the sensitivity of a previously described multilocus sequence typing (MLST) tool for T. vaginalis by using nested PCR. This enabled the typing of direct patient samples.
Participants From January to December 2014, we collected all T. vaginalis positive samples as detected by routine laboratory testing. Samples from patients either came from general practitioners offices or from the sexually transmitted infections (STI) clinic in Amsterdam. Epidemiological data for the STI clinic patients were retrieved from electronic patient files.
Primary and secondary outcome measures The primary outcome was the success rate of genotyping direct T. vaginalis positive samples. The secondary outcome was the relation between T. vaginalis genotypes and risk factors for STI.
Results All 7 MLST loci were successfully typed for 71/87 clinical samples. The 71 typed samples came from 69 patients, the majority of whom were women (n=62; 90%) and half (n=34; 49%) were STI clinic patients. Samples segregated into a two population structure for T. vaginalis representing genotypes I and II. Genotype I was most common (n=40; 59.7%). STI clinic patients infected with genotype II reported more sexual partners in the preceding 6 months than patients infected with genotype I (p=0.028). No other associations for gender, age, ethnicity, urogenital discharge or co-occurring STIs with T. vaginalis genotype were found.
Conclusions MLST with nested PCR is a sensitive typing method that allows typing of direct (uncultured) patient material. Genotype II is possibly more prevalent in high-risk sexual networks.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
Trichomonas vaginalis multilocus sequence typing (MLST) with nested PCR is a sensitive and culture independent typing method that can be used directly on patient material.
This is the first European study in which molecular epidemiology of T. vaginalis was performed using both sexually transmitted infection (STI) clinic samples and samples from general practitioners within one capital city.
With the exception of high-risk sexual behaviour, no epidemiological correlates were found with T. vaginalis genotype in an STI clinic population in Amsterdam, the Netherlands.
T. vaginalis MLST was performed on samples from one city in the Netherlands only.
The diversity using this MLST was so high that patients could not be linked directly but only two ‘genotypes’ could be discerned.
Introduction
Trichomonas vaginalis is globally the most common, non-viral, sexually transmitted pathogen and it is estimated that some 250 million new infections occur each year.1 T. vaginalis, an obligate extracellular protozoan, is the causative agent of trichomoniasis; a condition that can cause vaginitis in women and urethritis in men, but can also remain asymptomatic. Infection with T. vaginalis has been associated with increased risk of HIV acquisition.2
The publication of the T. vaginalis genome and recent advances in molecular typing methods have given scientists a better understanding of the genetic make-up of the organism, but have also provided the medical community a tool to study genetic relatedness between clinical samples.3 Such a tool potentially allows tracking of transmission routes, outbreaks and treatment failure versus reinfections. Despite these technical advances, only a few studies have looked into T. vaginalis genetic diversity in relation to patient demographic and clinical characteristics.4–7
Molecular typing studies consistently describe a two-type population structure for T. vaginalis, irrespective of the typing technique that was used, as reviewed in ref. 8. T. vaginalis genotype I proved to be more genetically diverse and, therefore, presumably older than genotype II in evolutionary history.8 Surprisingly, the two genotypes seem nearly equally prevalent across the globe, except for a preponderance for type I in South Africa and type II in Mexico.5 Of clinical relevance, type I is more frequently infected with the pathogenic T. vaginalis virus (TVV) and type II has been associated with metronidazole resistance.5 ,9
Previously, T .vaginalis has been typed using random amplified polymorphic DNA, restriction fragment length polymorphisms of a single gene, and 22 microsatellite markers, but these techniques are often difficult to reproduce in other laboratories.9–11 A more robust technique, multilocus sequence typing (MLST) was first introduced in 1998, as reviewed in ref. 12 and identifies polymorphism in approximately seven housekeeping genes through sequencing of PCR amplicons. Such an MLST was also developed for T. vaginalis.13 However, this MLST scheme requires culturing of patient material to ensure sufficient DNA load for testing.
Here we have modified the MLST method by improving sensitivity, using nested PCR, for the characterisation of T. vaginalis in clinical samples from the Public Health Laboratory in Amsterdam, the Netherlands. We aimed to identify molecular profiles for T. vaginalis and determine how these molecular profiles related to patient demographic and clinical characteristics.
Materials and methods
Trichomonas vaginalis clinical samples
Our study was performed at the Public Health Laboratory in Amsterdam, the Netherlands. We selected stored vaginal, urine or cervical samples from patients that had tested positive for T. vaginalis from January to December 2014. Samples were either collected as part of routine testing procedure at the STI outpatient clinic in Amsterdam, the Netherlands or were sent to the Public Health Laboratory by local general practitioners (GPs) for T. vaginalis testing. GPs and patients of the STI outpatient clinic are notified that samples sent to the Public Health Laboratory may be used for scientific research after having been anonymised. If patients object, their samples and clinical data are discarded.
T. vaginalis was detected using a transcription-mediated amplification (TMA) nucleic acid test (Hologic, Bedford, Massachusetts, USA). The remainder of the samples were stored at −20°C until further processing for this study. A data manager collected and anonymised demographic and clinical data from electronic patient files.
DNA extraction and amplification
Clinical samples used in this study were either cervical or vaginal swabs or first-catch urine that had been transferred directly into APTIMA transportation tubes (Hologic, Bedford, Massachusetts, USA). DNA was extracted from the APTIMA transportation tubes samples by isopropanol precipitation and the pellet was dissolved in 50 µL of 10 mM Tris/HCL.14 T. vaginalis positives in TMA were confirmed by a real-time T. vaginalis PCR previously described by Schirm et al,15 using Phocine herpesvirus DNA as internal control. We used previously published MLST primers to amplify the seven housekeeping genes: Tryptophenase (tryp), Glutaminase (glut), Family T2 aparaginase-like threonine peptidase (ft2a), Alanyl tRNA synthetase (alts), DNA mismatch repair protein (dmrp), Serine hydroxymethyltransferase (shmt) and Mannose 6-phosphate isomerase (m6pi).13 To increase the sensitivity of the MLST method, we amplified all loci by nested PCR. We used the published primers as inner primers and developed outer primers using gene sequences from the National Center for Biotechnology Information website. The inner and outer primers are listed in table 1. We modified the published dmrp reverse primer, as this primer overlapped with the trimming position. Inner primers were tagged with M13 and amplicons were sent to the Amsterdam Medical Center sequencing facility for further processing. Samples with low quality sequences were reamplified and sequenced at the Public Health Laboratory using an ABI 3130 genetic analyser (Applied Biosystems) as previously described.14
Multilocus sequence typing scheme for Trichomonas vaginalis
MLST typing
Consensus sequences were aligned using the MLST plugin in BioNumerics V.7 (Applied Maths, Belgium, Sint-Martens-Latem) and trimmed to the specified lengths (table 1). Only samples for which all loci could be sequenced were assigned a sequence type and used in further analyses. Allele numbers and sequence types (STs) were assigned according to STs previously published.13 Novel alleles and STs were numbered consecutively in order of discovery, following those previously published.13
Population structure and clustering analyses
We entered complete MLST profiles into the Bayesian clustering program STRUCTURE 2.3.3 using the admixture model with correlated allele frequencies.16 The program calculates probabilities for the number of populations (K) that the samples belong to according to allele frequencies at each locus. For each number assumed for K (ranging from K=1 to K=10), five runs of 105 iterations were performed, following a burn-in period of 105 iterations. The value for K best describing our population was derived using the ΔK method.17 ,18 MLST profiles were also entered into a minimum spanning tree using the MLST plugin of BioNumerics V.7 (Applied Maths, Sint-Martens-Latem, Belgium), using default settings. To compare our population structure with that of others using T. vaginalis MLST, we included the STs of 15 reference strains in our population structure analyses.13 ,19
Statistical analyses
All statistical analyses were performed in SPSS Statistics software V.21 (IBM). Differences in study population characteristics were tested using the χ2, Fisher-exact (when the expected value was <5) or Mann-Whitney U test, as appropriate.
Results
T. vaginalis clinical samples and study population
Approximately 3% (n=113/3804) of samples that were tested for T. vaginalis in 2014 were found positive (Public Health Laboratory yearly report 2014, http://www.ggd.amsterdam.nl/ggd/publicaties/jaarverslagen); 87 of these T. vaginalis positive samples, from 78 patients, were available for this study. Seventy-one (81.6%) of these samples, from 68 patients, were successfully typed for all 7 MLST loci and included in further analyses. Of the successfully typed samples, 34 came from 34 patients of the STI outpatient clinic and 37 samples came from 34 GP patients. One GP patient was sampled twice, 6 weeks apart and was included as two separate cases in the analyses. Two GP patients provided both a cervical and a urine sample at the time of testing; only one sample per patient was included in the analyses. Most study participants were women (61/68; 91%). Six of the 7 men included in the study were STI outpatient clinic patients. The median age of the total population was 31 years (IQR: 24.0–41.5); however, the STI clinic population was younger (median, 27 years (IQR: 23–33)) than the GP population (median, 35 years old (29–42); p=0.004). Demographic and clinical data were only available for the STI clinic population and these are listed in tables 2 and 3. The STI clinic population comprised mostly Surinamese (n=20; 61.8%); other ethnicities were: Dutch Antillean, North African and (non-Dutch) European (each n=3; 8.8%), Dutch (n=2; 5.9%) and finally Sub-Sahara African and Turkish (each n=1; 2.9%).
Demographic characteristics, by Trichomonas vaginalis genotype, of patients attending GP clinics and the STI outpatient clinic in Amsterdam, the Netherlands, from January to December 2014
Demographic and clinical characteristics, by Trichomonas vaginalis genotype, of patients attending the STI outpatient clinic in Amsterdam, the Netherlands, from January to December 2014
MLST profiles
We identified 6 previously described STs and 47 new STs (see online supplementary table S1). Of the 55 identified alleles, 14 had not been described before (table 1). Two patients provided a cervical and a urine sample at the time of testing and the MLST profiles were identical between samples from the same patient; this finding contributes to internal validation of the MLST profiling method. Using the current MLST method it was not possible to identify mixed infections.
supplementary table
T. vaginalis population structure
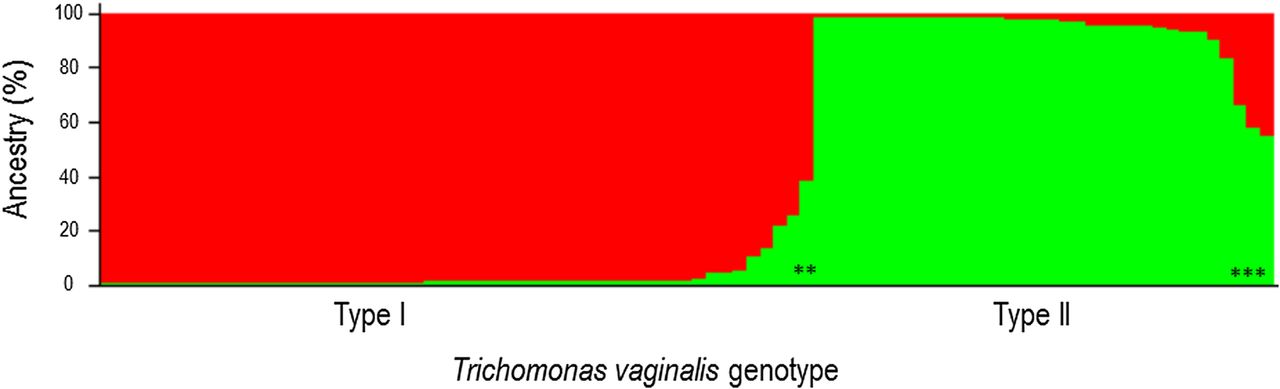
A two-type population structure was identified using the ΔK method on the probabilities calculated by STRUCTURE per assumed number of populations K (figure 1). Samples were assigned to a subpopulation if they shared at least 80% ancestry with that subpopulation. A two-population structure best fitted our data that resembled the type I and type II subpopulations previously described.10 ,13 ,19 STs of reference samples were included in our analyses to compare our population assignment with that of others.13 ,19 In this way we deduced that the larger subpopulation was type I and the smaller population was type II. Five samples (three reference and two clinical samples) shared <80% ancestry with the two populations and were not assigned (undetermined type I or II).

Assignment of clinical and reference samples by STRUCTURE. The larger subpopulation represents type I samples and the smaller subpopulation represents samples with a type II genotype. Five samples shared <80% ancestry with the two populations and were not assigned (undetermined type I or II) and are allocated with an asterix.
The minimum spanning tree (MST) also clearly displayed two major clusters and when we entered the STRUCTURE population allocations into the MST we found that the sample distribution overlapped for the two methods (figure 2). Interestingly, four of five samples that were not assigned to a population using STRUCTURE, clustered together with type I in the MST (figure 2). MLST sequence types per genotype are listed in online supplementary table S2.

{kind=link}
{kind=link}
Clustering of clinical and reference samples in a minimum spanning tree. Clustering and dissemination of the samples in the minimum spanning tree was identical to the subpopulations assigned by STRUCTURE (figure 1), with the exception of five samples, that were not assigned to a subpopulation by STRUCTURE (<80% ancestry), clustered together with type I samples in the minimum spanning tree. Grey partitioning shows clonal complexes within the population; samples that varied by only one locus. Branch lengths correspond to the number of loci that differed between connected nodes. Larger nodes indicate samples with identical MLST profiles. MLST, multilocus sequence typing.
supplementary table
T. vaginalis genotype and clinical and demographic characteristics of the patient
Two samples (one GP and one STI clinic patient's sample) shared <80% ancestry with either genotype and were excluded from the statistical analyses. Infections with T. vaginalis type I (n=40; 59.7%) occurred more frequently than type II (n=27; 40.3%) in our study population. Nonetheless, their distribution was equal for almost all clinical and demographic characteristics of the patients. The T. vaginalis genotype was not associated with age, ethnicity or clinical characteristics of the patient, or with urogenital symptoms such as vaginal or urethral discharge. The two genotypes were equally prevalent among patients from STI and GP clinics and among males and females (see tables 2, 3 and online supplementary figures). One significant association was noted between genotype and sexual behaviour: patients infected with type II reported more sexual partners in the preceding 6 months than patients infected with type I (p=0.028; see tables 2 and 3).
supplementary figure
Discussion
A previously published MLST method13 was successfully implemented to characterise T. vaginalis samples from Amsterdam, the Netherlands. By performing nested PCRs, we enhanced the MLST sensitivity and were able to use patient material that did not have to be cultured first. Similar to previous findings, two T. vaginalis subtypes emerged in our data and inclusion of STs from reference strains confirmed the previously described type I and II subpopulations.7 ,9–11 ,13 ,19 Apart from a positive association between type II and number of recent sex partners, no associations were seen between T. vaginalis genotype and patient characteristics.
T. vaginalis prevalence varies greatly per region and per population, with developing countries and minorities in developed countries being most at risk. Around 6% of women in Europe, compared to 18% of women in Africa, are estimated to harbour a T. vaginalis infection.20 African-American women are disproportionally affected by T. vaginalis with a prevalence of 13.3%, compared to only 1.3% of Caucasian Americans.21 Also, unlike other STIs, T. vaginalis prevalence is positively associated with age.1 ,22 Our study was not set up as a prevalence study, yet the patient demographics in our study likely reflect the distribution of T. vaginalis cases in Amsterdam. In our data, we also see that most cases were Surinamese—a minority population in the Netherlands originating from Suriname, a former Dutch colony. Most cases were women and most cases were older than 30 years. Prevalence data on men are scarce since they are not tested very often and prevalence in men is estimated to be eightfold lower than in women.20 The natural infection duration in men is unknown but is assumed to be no more than a few weeks, compared to months in women.23 Men are also not routinely tested for T. vaginalis at the STI clinic in Amsterdam and the six positive STI clinic male samples were detected in a male T. vaginalis and Mycoplasma genitalium screening study performed previously in our laboratory.24
Very few studies have described T. vaginalis genotype in relation to patient characteristics. Based on our current data it is unlikely that T. vaginalis genotype explains the ethnic or gender disparities that are commonly seen for T. vaginalis infection. T. vaginalis type I and II were equally distributed among patients of different ethnic origins, men and women and no association with T. vaginalis genotype and age was found. Conrad et al also found no association between T. vaginalis genotype and age.5 No previous studies have reported data on sexual risk behaviour in relation to T. vaginalis genotype and we now found that type II is positively associated with the number of reported sex partners in the preceding 6 months, possibly reflecting that this genotype circulates in high-risk sexual networks. Conrad et al assessed vaginal pH and positivity by Whiff test and found no association with T. vaginalis genotype.5 Brotman et al4 characterised the vaginal microbiota of T. vaginalis infected women and found T. vaginalis type I and II to be equally prevalent among women with different vaginal microbiota compositions, but this analysis included only seven women.
To date, the most striking association reported for T. vaginalis genotype is that type I seems to be more susceptible to metronidazole and is more frequently infected with TVV than type II.5 ,25 Conrad et al5 have shown that 73% of type I T. vaginalis strains were infected with TVV, compared with only 2.5% of type II strains. Four different genotypes for TVV have been described globally.26 TVV presence has been associated with symptoms and interestingly TVV genotype with severity of symptoms.26–28 In Cuba, TVV-1 was found only in mild to moderately symptomatic patients, whereas TVV-2 was only found in moderate to severely symptomatic patients.27 Previous studies that have studied the two-type T. vaginalis population structure in relation to metronidazole susceptibility have done so using the metronidazole minimum lethal concentration (MLC) method and have found a mean MLC for type I of 76.6 µg/mL and a mean MLC of 228.4 µg/mL for type II.5 ,25 Paulish-Miller et al25 have identified genetic markers in strains more resistant to metronidazole (SNPs leading to stop codons in genes encoding putative nitroreductases), but the authors emphasise that several mechanisms may be involved in metronidazole resistance. Nonetheless, information on metronidazole resistance is clinically relevant and has the potential to improve care in patients with treatment failure.5 ,29 Here we report only one GP patient that was sampled twice, 6 weeks apart, and we assume that the patient received metronidazole treatment between sampling. ST83 (T. vaginalis type II) was found in both samples, but, as we have no information on sexual behaviour; we do not know whether this was a persistent infection due to treatment failure or a reinfection.
A limitation of our study is that our sample size of patients with available clinical and demographic data was small and our population came from one (non-endemic) metropolitan region. We therefore cannot rule out that associations between T. vaginalis and patient characteristics may exist in other (endemic) regions. In this way, a bias for type I parasites (which are often infected with TVV) was seen in samples from South Africa, and moreover, a bias for type I parasites in HIV-positive women has also been reported.5 Those findings propose a possible parasite-virus-host interaction that needs to be investigated further, particularly in HIV-positive patients. No known HIV infections were present in our study population. As we worked with processed samples that could no longer be cultured, we did not set out to detect TVV and therefore do not know the prevalence or genetic make-up of TVV in our study population.
To conclude, the MLST profiling method for T. vaginalis is a robust typing method that can be reproduced between laboratories. By performing the MLST with nested PCRs we could use direct patient material that did not need to be cultured first. This method may allow easier collection and typing of specimens from high endemic, but low resource, regions. The molecular epidemiology of T. vaginalis is still in its infancy and further research is needed to fully understand transmission routes and pathogenicity of the parasite and its susceptibility to first choice drugs.
Acknowledgments
The authors wish to thank Martijn van Rooijen for providing anonymised patient data and Titia Heijman for coordinating specimen collection from the STI outpatient clinic. The authors also thank Maarten Schim van der Loeff for critically reviewing the manuscript.
References
Footnotes
Contributors CvdV and SMB conceptualised the study. MH and CvdV performed the laboratory work. CvdV drafted the manuscript. All authors reviewed and approved the final manuscript.
Funding This work was financially supported by the Public Health Service of Amsterdam (GGD Amsterdam), the Netherlands.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All MLST data are available in the online supplementary files. For additional data contact Sylvia Bruisten: sbruisten@ggd.amsterdam.nl