Article Text

Abstract
Introduction Finding therapeutic alternatives to carbapenems in infections caused by extended-spectrum β-lactamase-producing Escherichia coli (ESBL-EC) is imperative. Although fosfomycin was discovered more than 40 years ago, it was not investigated in accordance with current standards and so is not used in clinical practice except in desperate situations. It is one of the so-called neglected antibiotics of high potential interest for the future.
Methods and analysis The main objective of this project is to demonstrate the clinical non-inferiority of intravenous fosfomycin with regard to meropenem for treating bacteraemic urinary tract infections (UTI) caused by ESBL-EC. This is a ‘real practice’ multicentre, open-label, phase III randomised controlled trial, designed to compare the clinical and microbiological efficacy, and safety of intravenous fosfomycin (4 g/6 h) and meropenem (1 g/8 h) as targeted therapy for this infection; a change to oral therapy is permitted after 5 days in both arms, in accordance with predetermined options. The study design follows the latest recommendations for designing trials investigating new options for multidrug-resistant bacteria. Secondary objectives include the study of fosfomycin concentrations in plasma and the impact of both drugs on intestinal colonisation by multidrug-resistant Gram-negative bacilli.
Ethics and dissemination Ethical approval was obtained from the Andalusian Coordinating Institutional Review Board (IRB) for Biomedical Research (Referral Ethics Committee), which obtained approval from the local ethics committees at all participating sites in Spain (22 sites). Data will be presented at international conferences and published in peer-reviewed journals.
Discussion This project is proposed as an initial step in the investigation of an orphan antimicrobial of low cost with high potential as a therapeutic alternative in common infections such as UTI in selected patients. These results may have a major impact on the use of antibiotics and the development of new projects with this drug, whether as monotherapy or combination therapy.
Trial registration number NCT02142751. EudraCT no: 2013-002922-21. Protocol V.1.1 dated 14 March 2014.
- BACTERIOLOGY
- GENITOURINARY MEDICINE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
The investigation of fosfomycin efficacy as monotherapy in bacteraemic urinary tract infections by Escherichia coli is justified.
-
The new proposed paradigms for investigating new alternatives for antibiotic-resistant bacteria through randomised clinical trial designs in order to meet the real clinical needs were considered for this study design.
-
This clinical trial is proposed as an initial step in the investigation of a low cost, orphan antimicrobial with a high potential as a therapeutic alternative for frequent infections caused by multidrug-resistant E. coli in selected patients.
-
The results may have a major impact in the use of antibiotics and in the development of new projects with this drug, both in monotherapy or in combination therapy.
-
The open-label design is theoretically more prone to bias; however, we use a remote automatic randomisation system after collection of baseline data, hard outcomes as secondary variables and external evaluation by blinded investigators.
Background
The scarcity of available drugs for the treatment of infections caused by multidrug-resistant (MDR) and extensively drug-resistant pathogens is recognised as a public health problem. Besides the efforts on infection control or facilitating and promoting new drug development,1 old drugs may offer some solutions in the short term. On one hand, some old drugs may be active against some MDR pathogens, offering an alternative for therapy in desperate situations. On the other hand, old drugs may have avoided overuse, unlike other broad-spectrum antibiotics, thus contributing to limit the selective pressure posed by the latter, which facilitates the spread of emerging resistant bacteria. However, because of real, urgent medical needs, some of these old drugs are being used without solid evidence. High-quality clinical research in the MDR field is challenging;2 this is particularly true in the case of old drugs because studies must typically be designed and driven by academic investigators.
Extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae and particularly Escherichia coli have been established in the last decade as a common cause of infection worldwide.3 Since carbapenems are considered the drugs of choice for serious infections caused by these microorganisms, consumption of these drugs is increasing,4 which is contributing to the selection and spread of carbapenem-resistant Gram-negative bacilli.5
In this setting, therapeutic alternatives to carbapenems for the treatment of ESBL-producing Enterobacteriaceae are urgently needed. Since these organisms are usually resistant to penicillins, cephalosporins and quinolones, the most plausible alternatives are β-lactam/β-lactamase inhibitor combinations, temocillin (available only in a few countries), aminoglycosides (the limitations of which are well known6) and fosfomycin. Fosfomycin, an antibiotic discovered more than 40 years ago, acts by inhibiting the formation of peptidoglycans during the bacterial cell wall biosynthesis. This antibiotic is frequently active against MDR and extremely resistant Enterobacteriaceae,7 and in particular against ESBL-EC.8
Fosfomycin, in its intravenous formulation (disodium fosfomycin), is approved in Spain, according to a summary of product characteristics (SCP), for clinical use in a wide variety of infections caused by susceptible organisms, including complicated urinary tract infections (UTI) and urinary sepsis (in this case it is advised to be used in combination with other active drugs).9 The recommended dose is 4 g every 6–8 h. It is important to consider that when this drug was developed, the requirements of the regulatory agencies were different from those currently applicable.
A systematic review on the effectiveness of fosfomycin in the treatment of infections caused by MDR Enterobacteriaceae10 was published in 2010; the existing information on the efficacy in systemic infections caused by these microorganisms was virtually non-existent; moreover, they were limited to a few small series of cases in which they were used in combination with other active drugs. Thereafter, not much information has been generated.
The objective of this article is to describe the hypothesis, objectives, design, variables and procedures for a randomised controlled trial with fosfomycin.
Methods/design
The FOREST study is a phase 3, randomised, controlled, multicentric, open-label clinical trial to prove the non-inferiority of fosfomycin versus meropenem in the targeted treatment of bacteraemic UTI due to ESBL-EC, designed as a real practice trial. It is a non-commercial, investigator-driven clinical study funded through a public competitive call by Instituto de Salud Carlos III, Spanish Ministry of Economy (PI13/01282). The study is coordinated by investigators from Hospital Universitario Virgen Macarena in Seville, Spain; the sponsorship is performed by Fundación Pública Andaluza para la Gestión de la Investigación en Salud de Sevilla (FISEVI), of which the sponsor-scientific responsibilities are delegated to the CTU (Clinical Trial Unit—Hospital Universitario Virgen del Rocío, Seville, Spain).
All participating patients or their relatives must give written informed consent before any study procedures occur, including the withdrawal of biological samples for the study. Informed consent form and patient information sheet are included as online supplementary appendix 1.
Study hypothesis and objectives
The hypothesis to test is that intravenous fosfomycin is not inferior to meropenem for the targeted treatment of bacteraemic UTI caused by ESBL-EC in terms of efficacy. The primary objective of the study is to demonstrate that intravenous fosfomycin is not inferior to meropenem for reaching clinical and microbiological cure 5–7 days after the completion of treatment. Secondary objectives include comparing the early clinical and microbiological response, 30-day mortality, hospital stay, recurrence rate, safety and impact on intestinal colonisation by MDR Gram-negative bacilli, evaluation of the rate of resistance development to fosfomycin and blood level concentration of fosfomycin. The outcome definitions and time frames on which they are measured are described in table 1.
Description of outcome variables and time frames
Selection and enrolment
Hospitalised adults (18 years of age or older) with bacteraemic UTI caused by fosfomycin and meropenem susceptible ESBL-EC are candidates to be included in the study. Eligible patients will be detected from the daily review of blood culture results. Inclusion and exclusion criteria are detailed in table 2. The setting for the study will be 22 public and academic hospitals with research groups pertaining to the Spanish Network for Research in Infectious Diseases (REIPI) and/or the Spanish Study Group of Nosocomial Infections (GEIH) of the Spanish Society of Infectious Diseases and Clinical Microbiology (SEIMC).
Inclusion and exclusion criteria
Randomisation
A 1:1 randomisation system allows the assignment of treatment arms, either fosfomycin or meropenem. Randomisation is stratified according to active or non-active previous empirical treatment received for the infection in order to ensure the homogeneous distribution of empirical active treatment. The automatic randomisation system allows the inclusion of patients 24 h a day, 7 days a week, and is integrated in the electronic case report form (e-CRF) of the study. A copy of the randomisation list is in the CTU, so in case of technical problems, data can be easily reached and further randomisation allowed.
Trial intervention and control
Each patient will enter one of the following treatment branches:
-
Study arm A: intravenous disodium fosfomycin 4 g/intravenously/6 h in 60 min infusion.
-
Study arm B: intravenous meropenem 1 g/intravenously/8 h in 15–30 min infusion.
Switch to oral therapy is allowed from the fifth day of treatment with the study medication to complete 10–14 days of therapy if all the following conditions are fulfilled: clinical improvement has been achieved, there is haemodynamic stability, the patient can tolerate oral intake and the isolate is susceptible to one of the following options. For patients in arm A, intravenous fosfomycin can be switched to oral fosfomycin trometamol 3 g/48 h.
For patients in arm B, intravenous meropenem can be switched to one of the following oral drugs in the specified sequence, based on the susceptibility tests.
-
Ciprofloxacin 500 mg/12 h
-
Amoxicillin/clavulanate 500/125 mg/8 h
-
Trimethoprim-sulfamethoxazole 160/800 g/12 h
Treatment assignment is intended for targeted treatment of bacteraemic UTI caused by ESBL-EC. Concomitant treatment with any other systemic antibiotic with intrinsic activity against Gram-negative bacilli is not permitted. The administration of any of these drugs while the patient is receiving the study drug will be deemed as a withdrawal criterion.
Considering that all the study drugs are officially approved for urinary tract sepsis in Spain, the sponsor will not provide the study drugs; permission for the use of the drugs through the normal provision of each Pharmacy Hospital has been obtained for every site participating in the study. In order to ensure the tracking of the products administered, the lot number and expiration dates will be recorded. This is also required by the Spanish Regulatory Agency.
Dose adjustment is detailed in case of renal dysfunction for all study drugs according to creatinine level clearance as described in table 3. For this reason, renal function is monitored during the entire duration of antibiotic treatment.
Dose adjustment according to renal functioning
There are no absolute contraindications for the use of any other drugs during the study. However, contraindications, warnings and precautions for their use and possible interactions with the study drugs are to be taken into account. Only drugs used as a consequence of adverse events will be collected in the e-CRF of the study.
Follow-up scheme
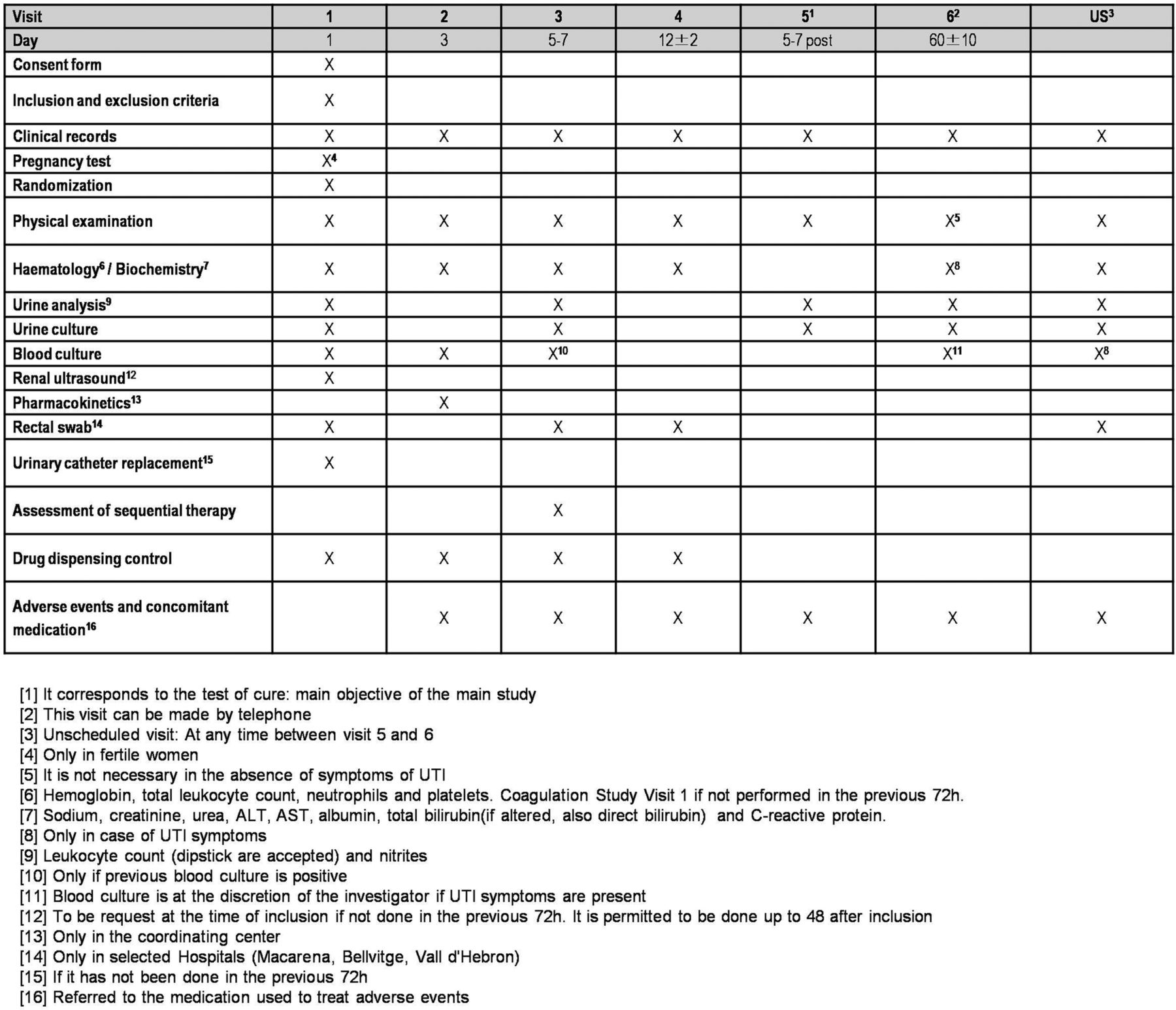
Patients included in the study have to be followed for 60 days (±10 days) after the diagnosis of bacteraemic UTI. Follow-up will be organised in six planned visits as: V1, baseline or day 1; V2, day 3; V3, day 5–7; V4, end of treatment or day 12 (±2 days); V5, 5–7 days after treatment completion (test of cure); and V6, day 60 (±10 days). Additionally, data from unplanned visits will be collected with special consideration for the occurrence of any adverse event or recurrence. A flow chart for the study is included in figure 1. Procedures to be performed during those visits are specified in figure 2. The visit schedule is planned in order to obtain data for clinical status, samples collection, and efficacy and safety variables, including renal and liver monitoring functioning, and adverse events. At the final evaluation up to 60 days of follow-up, data for all the outcome variables will be gathered.

FOREST study flow chart (PK, pharmacokinetics; UTI, urinary tract infection; ESBL-E. coli, extended-spectrum β-lactamase-producing Escherichia coli).
{kind=link}
{kind=link}
Schedule of visits and assessments (ALT, alanine transaminase; AST, aspartate aminotransferase; UTI, urinary tract infection).
Biological samples
All the sites are asked to locally process the blood and urine cultures at the times described in the schedule of visits, using standard microbiological techniques for the isolation and identification of bacteria; the microbiology laboratories of these centres use the Quality Control system of the Spanish Society of Infectious Diseases and Clinical Microbiology (SEIMC). Susceptibility test are to be interpreted according to EUCAST recommendations. The isolated ESBL-EC are to be sent to the central laboratory located in Hospital Universitario Virgen Macarena in Seville in order to confirm the identification, ESBL production, susceptibility testing using reference techniques and ESBL characterisation through PCR and sequencing.
Three hospitals (Hospital Universitario de Bellvitge, Hospital Universitario Vall d'Hebrón, both in Barcelona, and Hospital Universitario Virgen Macarena, Seville) will participate in the study of the rectal carriage of ESBL-producing and carbapenemase-producing Gram-negatives by taking rectal swabs from participants at different times, as set out in the schedule of visits; 60 patients are expected to be included in the rectal carriage study. Additionally, fosfomycin serum concentrations will be measured in a sample size of 20 patients at only one site (Hospital Universitario Virgen Macarena).
All study samples will be anonymised, being identified only by the patient study code, in order to ensure that the association with personal data is not possible. The objective and management of these samples are included in the patient information sheet and informed consent form. Specific details for sample management and procedures are included in online supplementary appendix 2.
Outcome measures
The primary efficacy end point is the clinical and microbiological cure at 5–7 days after finalisation of treatment (test of cure, TOC); it will be assessed in the modified intention-to-treat (m-ITT) population, formed by all patients with evaluable microbiological diagnosis of bacteraemic UTI due to ESBL-EC who received at least one dose of intravenous antibiotics. The primary efficacy end point will be evaluated by blinded investigators.
Secondary end points will include early clinical response, early microbiological response, length of hospital stay, impact of study treatment in the colonisation by MDR Gram negatives, relapse rate and reinfection rate, emergence of E. coli resistant to fosfomycin or meropenem, fosfomycin steady-state plasma concentrations, safety of intravenous fosfomycin in this indication and mortality of any cause for the complete follow-up period (table 1).
Safety will also be evaluated in the m-ITT population. Any adverse event occurring from the informed consent form signature to 28 days after the last dose of study medication will be recorded.
Sample size calculation
The sample size calculation was performed on the basis of 80% power to reject null hypothesis with a two-tailed significance level of 5%. Assuming estimated clinical cure rates of 90% in the control group and 85% in the experimental group, for non-inferiority margin of 7%, with an assignment in 1:1 proportion and 5% of lost patients, a total of 198 patients (99 patients in each group) were needed.
Participating centres were selected after a feasibility survey in which data for bacteraemic UTI by ESBL-EC in 2012 were included. All participating centres were committed to include at least 10 patients in the study period, competitively, thereby achieving the guaranteed sample size. Time schedule of enrolment is 24 months from the first patient inclusion.
Statistical analysis
The absolute difference, and 95% CI in clinical and microbiological cure rate at TOC between patients in both arms, will be calculated. Multivariate analysis using logistic regression for the main outcome will be performed in order to ensure the independence of the effect of treatment. Special consideration will be taken in the multivariate analysis considering the site origin of the study sample. Absolute difference with 95% CI in early clinical cure rates and early microbiological cure (5th day of treatment), mortality, rate of adverse events and rectal colonisation with antibiotic-resistant Gram negatives will also be calculated. Also, average hospital stay will be compared between both study arms.
A description of the fosfomycin plasma concentration and its variability among patients will be performed.
Interim analysis
An interim analysis has been planned to be carried out when 50% of cases are recruited. This analysis will include a safety assessment performed by an independent committee of three experts pertaining to REIPI network but not participating in this study.
Definition of analysis population
ITT: all randomised patients.
m-ITT: randomised patients who have received at least one dose of intravenous antibiotics.
Clinically evaluable population (CE): patients who have completed 5 days of intravenous (or who have died after having received at least one dose of intravenous antibiotics) and a total duration of at least 10 days, with at least 75% of the total amount of oral antibiotics if treatment was performed sequentially.
Clinical and microbiologically evaluable population (CME): the population clinically evaluable in whom microbiological tests (blood and urine cultures where applicable) at the indicated follow-up visits were performed.
Safety and adverse event reporting
Pharmacovigilance activities including the registration, reporting and communication of all adverse events occurred within the patients included in the clinical trial are mandatory as part of the legal requisitions applicable in clinical trials. For this reason, the responsibility of performing those activities was derived from the sponsor to the CTU.
In order to recollect all the information related with possible adverse events in the study, every study team is trained during the site initiation visit on the definitions of adverse events and rules for communication. Any adverse event related, or not with the study medication, has to be gathered in the e-CRF, which contains a specific pharmacovigilance module. Serious adverse events (SAE) are mandatorily to be completed with more detailed information comprising SAE description (according to international dictionaries in pharmacovigilance), date of onset and resolution, severity, assessment of causality to study medication, action taken and other concomitant medication/procedures. Any adverse event occurred is followed by initial and follow-up/s communication/s until resolution.
The crucial data related to the adverse event are to be filled in a specific form provided for the study. The SAEs form is centralised in the CTU, the personnel of which are responsible for the reception (by fax or email communication), and registering and resolution of queries to the sites. The identification of any Serious Unexpected Adverse Event (SUSAR) is assessed by a safety medical monitor in order to evaluate if the information is to be communicated to Regulatory Authorities, Ethics Committees and Investigators following Good Clinical Practices (GPCs) rules. In that case, communication through the EudraVigilance system is foreseen. Safety annual reports are issued with all the safety information in the study being reported to regulatory Authorities and Ethics Committees. The safety medical monitor is responsible for any update in safety information of the investigational medicinal product (IMP).
Study organisation
The study coordinating team (SCT) is formed by the scientific group in the coordinating site (Hospital Universitario Virgen Macarena) and the Clinical Trials Unit in Hospital Universitario Virgen del Rocío, the personnel of which are responsible for the entire coordination of the study in the sites involved. These personnel will submit the administrative authorisations for the study, handle regulatory affairs, provide ethics committee contact and response, take up safety monitoring and pharmacovigilance responsibilities of the sponsor, as well as provide logistic coordination and a contact point for all the 22 participating hospitals.
CTU acts as delegation figure of the sponsor (FISEVI, managing foundation for research in Seville) in relevant activities evolved in a multicentre trial. Monitoring activities in Spain are performed by clinical research associates (CRAs) connected with the Spanish Clinical Trial Network in public hospitals. CTU is in close contact with the scientific coordination of the study, acting accordingly and in a parallel manner, so that necessary decisions have been taken after previously having consulted with the study coordination team (SCT). All efforts will be made in order to maintain the recruitment rhythm needed for achieving the sample size through continuous communication with the participant sites.
Data and safety monitoring
The quality of all data collected will be carefully supervised by the CTU; individual responsible for the revision and update of data collection will be in close contact with the investigators, in order to perform a close follow-up of the study procedures, data update and corrections through email or phone contact. Beside this, visits will be organised in order to perform data source verification according to a monitoring plan.
An independent, objective review of all accumulated data from the clinical trial is foreseen. This will be performed at the time of the interim analysis when 50% of the sample has been included. Based on this review, the independent committee (3 independent investigators from REIPI) will advise the sponsor on the appropriateness of continuing the clinical trial as designed.
Ethical considerations
We do not consider having any special ethical considerations beyond those typical for the development of a randomised trial. The study will be carried out according to the principles of the Declaration of Helsinki, and according to the legal norm directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of Good Clinical Practice in the conduct of clinical trials on medicinal products for human use.
The trial was started after obtaining approval of an Ethics Review Committee, conformity of the Directors of the Institutions, and the authorisation of the Spanish Regulatory Agency (AEMPS, Agencia Española del Medicamento y Productos Sanitarios) and the Institutional Review Boards (IRBs) at each site participating in the trial. A formal contract agreement was signed with each of the institutions and with the sponsor of the study. Any modification to the protocol that could impact on the conduct of the study or potential benefit of the patient, or that may affect patient safety, including changes of study objectives, study design, patient population, sample size, study procedures or significant administrative aspects, will require a formal amendment to the protocol. Such amendment will be agreed on by the study coordinating team (CTU will act as delegation figure of the sponsor) and will be approved by IRBs prior to implementation and notified to the health authorities in accordance with local regulations.
The confidentiality of records that could identify subjects in the FOREST study will be protected in accordance with the EU Directive 2001/20/EC. All the laws that legislate for the control and protection of personal information will be carefully followed. The identity of patients will not be disclosed in the e-CRF; names will be replaced by an alphanumeric code and any material related to the trial as samples will be identified in the same way so that any personal information can be revealed.
SPIRIT 2013 Explanation and Elaboration paper and SPIRIT statement11 ,12 will be followed for the reporting of results to any scientific journal or event.
Discussion
Fosfomycin has been identified as an orphan antibiotic with high potential value in order to be investigated in this era of antibiotic resistance.13 No studies have been found on fosfomycin for the proposed or similar indication in careful revision of the clinical trials public registries. Therefore, an opportunity to design and conduct a randomised clinical trial to evaluate its efficacy and safety presented itself. In this sense, it seems especially important to take into consideration the new proposed paradigms for investigating new alternatives for antibiotic resistant bacteria through randomised clinical trial designs in order to meet the real clinical needs.14 ,15
The main concern with the use of fosfomycin is the possibility of resistance development during treatment. While it is true that spread of resistant strains in our environment has been linked to increased consumption of drugs for oral treatment of uncomplicated UTI,16 it seems that the resilience of fosfomycin-resistant enterobacteria has decreased, which would otherwise have permitted it to maintain its activity over time.17 Even though development of resistance to fosfomycin can occur during treatment, it seems to be much less frequent in E. coli than in Klebsiella spp or Pseudomonas aeruginosa, and specifically in UTI,17 which provides an adequate background for testing the efficacy of this drug in monotherapy for bacteraemic UTI caused by ESBL-EC. Also, bacteraemic infections with a source in the urinary tract, especially in the absence of urine obstruction, is associated with lower mortality in comparison with other sources;18 finally, the development of resistance in the course of treatment is less likely in these infections.17 Therefore, the investigation of fosfomycin efficacy as monotherapy in bacteraemic UTIs by E. coli, at least if there has been an adequate source control if necessary, is justified.
The few pharmacokinetic and pharmacodynamic studies performed to date with fosfomycin indicate that sufficient plasma concentrations are reached for the treatment of systemic infections due to susceptible organisms for at least 4 h19 ,20 following an intravenous dose of 4 g. Since it is excreted unchanged in the urine, its levels in the urine are also suitable for diagnosing these infections.19 Pharmacokinetic data available to date have been obtained following administration of single doses;21 however, we intend to determine fosfomycin plasma levels following repeated doses (48 h after treatment).
The Food and Drug Administration (FDA) considers ‘complicated UTI’ to be a syndrome for new therapy evaluation.22 However, we believe the syndrome definition of the FDA is overly heterogeneous as it includes different clinical situations, from lower UTI in catheterised patients, to pyelonephritis with bloodstream infection in patients with structural of functional problems in the urinary tract. The research group responsible for this trial considers that using such definitions for investigating fosfomycin would not provide valuable results, particularly for patients with bacteraemia; furthermore, the results can be readily transferable to non-bacteraemic UTIs. This is why we decided to include patients with BUTI, who are readily identifiable and for whom clinical decisions are taken every day in real practice.
We decided to use meropenem as comparator because carbapenems are considered the drugs of choice for invasive infections caused by ESBL producers,23 and there is extensive experience with meropenem. Ertapenem was not considered because treatment of UTIs is not an approved indication for this drug in Europe.24
Switching to oral therapy was considered in our study, with the aim of reflecting clinical practice. However, decisions were not easy at this point. Fosfomycin trometamol is an oral formulation of fosfomycin reaching low plasma concentrations but very high urinary concentrations;18 the results from observational studies suggest that fosfomycin trometamol is useful for the treatment of cystitis and complicated UTIs caused by ESBL-EC.10 ,25 Anecdotal experience from our team with the use of fosfomycin trometamol in this situation has been positive (Rodríguez-Baño J, unpublished data). Therefore, once the bacteraemia and source of infection have been controlled, which is the objective in the first phase of intravenous treatment, the use of oral fosfomycin trometamol is reasonable. For the control arm, and since there are no oral carbapenems available, we needed to look for other alternatives. Because the susceptibility of ESBL-EC to oral drug is heterogeneous, we defined a step-based strategy. Because of the extensive experience with fluoroquinolones in these infections, ciprofloxacin is our first option; however, most ESBL-EC are resistant. The second option is amoxicillin/clavulanate, which is active against a significant proportion of ESBL-EC and has been shown to be effective in observational studies.25 ,26 Finally, trimethoprim-sulfamethoxazole is also recommended for pyelonephritis caused by susceptible strains and is, therefore, included as the third option.
Regarding the safety of disodium fosfomycin, available data suggest it is a very well tolerated drug.10 Because the sodium content is high (330 mg of sodium per gram of fosfomycin), the Spanish Medicines Agency recommends to take this into account in patients requiring sodium restriction,9 and therefore we excluded patients with moderate-to-severe heart insufficiency, liver cirrhosis or renal impairment receiving dialysis. Also, determination of plasma sodium and presence of oedema are incorporated in the follow-up visits. However, more data from well-designed studies are clearly needed.
This clinical trial is proposed as an initial step in the investigation of a low cost, orphan antimicrobial with a high potential as a therapeutic alternative for frequent infections caused by MDR E. coli in selected patients. The results may have a major impact in the use of antibiotics and in the development of new projects with this drug, both in monotherapy or in combination therapy.
Trial status
-
Funding for the study was approved on November 2013 and available for study expenses in January 2014.
-
Authorisation from the Spanish Regulatory Authority was obtained on 5 May 2014.
-
Approval for the EC for the 22 sites included was obtained on 27 July 2014.
-
A total of 15 of 22 sites have been officially opened at the time of manuscript submission.
-
First patient inclusion for the study occurred on 1 August 2014.
-
Study is approved until August 2017 (recruitment period 2 years).
-
Dissemination of results directed to patients will be channelled through the Spanish Clinical Studies Registry (Agencia Española del Medicamento y Productos Sanitarios), of which content is adapted to patients.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online appendix 1
- Data supplement 2 - Online appendix 2
Footnotes
-
Collaborators FOREST Study Team—JR-B, Virgen Macarena Hospital Universitario, Seville; Dr Natera Kindelán, Hospital Universitario Reina Sofía, Córdoba; Dr Salavert Lletí, Hospital Universitario La Fe, Valencia; Dr Merino de Lucas, Hospital Universitario General de Alicante; Dr Amador Prous, Hospital Marina Baixa, Alicante; Dr Hernández Torres, Hospital Universitario de la Arritxaca, Murcia; Dr Shaw Perujo, Hospital Universitario de Bellvitge, Barcelona; Dr Pomar, Hospital Universitario Santa Creu e Sant Pau, Barcelona; Dr Calvo Sebastián, Hospital Universitario de Terrassa, Barcelona; Dr Sorli Redó, Hospital del Mar, Barcelona; Dr Pigrau Serrallach, Hospital Universitario Vall d'Hebrón, Barcelona; Dr Barcenilla Gaite, Hospital Universitario Arnau de Villanova, Lleida; Dr Sequeira Lopes da Silva, Hospital Universitario 12 de Octubre, Madrid; Dr Pintado García, Hospital Universitario Ramón y Cajal, Madrid; Dr Martínez Martínez, Hospital Universitario Marqués de Valdecilla, Santander; Dr Fleites Gutierrez, Hospital Universitario Central de Asturias; Dr Bereciartua Bastarrica, Hospital Universitario de Cruces, Vizcaya; Dr Dueñas Gutiérrez, Hospital Universitario de Burgos; Dr Martínez Álvarez, Hospital Royo Villanova, Zaragoza; Dr Borrell Solé, Complejo Universitario Son Espases, Balearic Islands; Dr Cárdenas Santana, Hospital Universitario Gran Canaria, Dr Negrín, Gran Canaria; and Dr Lecuona Fernández, Hospital Universitario de Canarias, Santa Cruz de Tenerife.
-
Contributors JR-B and JS-D were responsible for formulating the overall research questions and for the methodological design of the study. CR-F collaborated in the submission of the project for the Spanish funding, wrote the first draft of the manuscript and also collaborated in the methodological aspects of the study. JR-B is the coordinating investigator and leader of the Coordination Team. CR-F is responsible for the CTU and the pharmacovigilance monitor, and AB and LL-A collaborated in the organisation of the study. IL-H and AP contributed in all the microbiological details of the study. VM and MC are responsible of the fosfomycin pharmacokinetics study. JR-B and JS-D participated in its design and supervised the project. All authors read and approved the final manuscript.
-
Funding FOREST study is a project funded through public competition by the Spanish Government, Ministerio de Economía y Competitividad, Instituto de Salud Carlos III (PI13/01282)—co-financed by European Development Regional Fund ‘A way to achieve Europe’ ERDF, Spanish Network for the Research in Infectious Diseases (REIPI RD12/0015). Sponsorship of the study is performed by Fundación Pública Andaluza para la Gestión de la Investigación en Salud de Sevilla (FISEVI), a public research managing foundation, of which the sponsor scientific responsibilities are delegated to the CTU.
-
Competing interests JR-B has been the speaker for Pfizer, Merck, Gilead and AstraZeneca, has received unrestricted grants for research from Novartis, and has served as scientific consultant for Merck, AstraZeneca and Roche.
-
Patient consent Obtained.
-
Ethics approval Ethical approval for this trial, which is in compliance with the Helsinki Declaration and in agreement with the SPIRIT statement, was obtained for the authorisation of the Spanish Regulatory Authority and the Coordinating Institutional Review Board (IRB) of Biomedical Research in Andalusia (Referral Ethic Committee), which gathered the approval from local ethic committees in all the participating sites in Spain (22 sites).
-
Provenance and peer review Not commissioned; internally peer reviewed.