Article Text

Abstract
Objective To investigate the effect of regular paracetamol on bronchial hyper-responsiveness (BHR) and asthma control in adult asthma.
Setting Single research-based outpatient clinic.
Participants 94 adults with mild-to-moderate asthma received randomised treatment; 85 completed the study. Key inclusion criteria were age 18–65 years, forced expiratory volume in 1 s (FEV1) >70% predicted, provocation concentration of methacholine causing a 20% reduction in FEV1 (PC20) between 0.125 and 16 mg/mL. Key exclusion criteria included an asthma exacerbation within the previous 2 months, current regular use of paracetamol, use of high-dose aspirin or non-steroidal anti-inflammatory drugs, current or past cigarette smoking >10 pack-years.
Interventions In a 12-week randomised, double-blind, placebo-controlled, parallel-group study, participants received 12 weeks of 1 g paracetamol twice daily or placebo twice daily.
Primary and secondary outcome measures The primary outcome variable was BHR, measured as the PC20 at week 12. Secondary outcome variables included FEV1, fractional exhaled nitric oxide (FeNO) and asthma control questionnaire (ACQ) score.
Results At 12 weeks, the mean (SD) logarithm base two PC20 was 1.07 (2.36) in the control group (N=54) and 0.62 (2.09) in the paracetamol group (N=31). After controlling for baseline PC20, the mean difference (paracetamol minus placebo) was −0.48 doubling dose worsening in BHR in the paracetamol group (95% CI −1.28 to 0.32), p=0.24. There were no statistically significant differences (paracetamol minus placebo) in log FeNO (0.09 (95% CI −0.097 to 0.27)), FEV1 (−0.07 L (95% CI −0.15 to 0.01)) or ACQ score (−0.04 (95% CI −0.27 to 0.18)).
Conclusions There was no significant effect of paracetamol on BHR and asthma control in adults with mild-to-moderate asthma. However, the study findings are limited by low power and the upper confidence limits did not rule out clinically relevant adverse effects.
Trial Registration Australia New Zealand Clinical Trials Registry Number: NZCTR12609000551291.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
Randomised placebo-controlled trial.
-
Physiological, clinical and immunological outcome measures.
-
Powered to detect a marked effect on BHR.
Introduction
There is a growing body of evidence to suggest that paracetamol may play an important role as a risk factor in the development of asthma, and that increasing worldwide use may have contributed to the increasing global prevalence of asthma observed over the past 40 years.1 ,2 Childhood asthma risk increases in the offspring of women who consume paracetamol during pregnancy,3 and paracetamol use in the first 12 months of life is associated with an increased risk of wheezing at 3 years4 ,5 and 6–7 years.6 Cross-sectional surveys in children,6 adolescents7 and adults8–11 consistently demonstrate an association between current paracetamol use and asthma in populations with widely differing lifestyles, standards of living, medical practice and availability of paracetamol. However, there is also evidence that these associations may, in part, be due to confounding by indication in some,12–14 but not all cohort studies in childhood.15 Cohort studies in adults have demonstrated that increasing frequency of paracetamol use is positively associated with newly diagnosed (adult-onset) asthma.16 ,17
Evidence also indicates that paracetamol may increase the severity of asthma in those with the disease. This primarily comes from the only randomised controlled trial of the effect of paracetamol use for fever and asthma outcomes, in which asthmatic children experiencing a current febrile illness were randomised to receive either paracetamol or ibuprofen.18 Children who received paracetamol were more likely to require an outpatient visit for asthma compared with children in the ibuprofen group. The increased risk with paracetamol was dose dependent and related to respiratory febrile illnesses rather than other causes of fever. In a case–control study, which reported a dose-dependent association between paracetamol use and asthma, a progressively greater risk in those with more severe disease was noted, suggesting an effect on causation and severity of the disease.10
The mounting epidemiological evidence, supported by several biologically plausible mechanisms19–28 has led to repeated calls2 ,5–7 ,13 ,29–32 for randomised controlled trials to be undertaken to explore the relationship between paracetamol and asthma. This study is the first randomised placebo-controlled trial undertaken to investigate the effect of regular daily paracetamol on asthma severity in adult patients with asthma. It was powered to detect a one doubling dose change in PC20 methacholine bronchial hyper-responsiveness (BHR). Markers of airways inflammation and systemic immunological responses were monitored to provide insight into possible mechanisms of action. The hypothesis was that regular paracetamol use would result in a worsening in BHR and asthma control.
Methods
The study design was a double-blind, randomised, placebo-controlled, parallel group trial based in Wellington, New Zealand. The study methods are summarised with additional details provided in the online supplementary appendix.
Participants
Participants were identified from the Medical Research Institute of New Zealand (MRINZ) asthma register, general practitioner patient databases and the general public through advertising. Inclusion criteria included age between 18 and 65 years, wheeze in the previous 12 months and a doctor's diagnosis of asthma, forced expiratory volume in 1 s (FEV1) ≥70% predicted at screening and baseline and a PC20 MCh (the provocation concentration of methacholine causing a 20% reduction in FEV1) of between 0.125 and 16 mg/mL at baseline. Exclusion criteria included regular use of theophylline, ipratropium bromide, tiotropium or leukotriene receptor antagonists in the previous 3 months, alanine aminotransferase (ALT) levels greater than 1.5 times the upper limit of normal at baseline, a history of liver disease or the current use of hepatotoxic drugs, an exacerbation of asthma within the previous 2 months requiring prednisone or nebulised bronchodilator, current or past cigarette smoking >10 pack-years, history of sensitivity or allergy to paracetamol or current regular use of paracetamol, use of high-dose aspirin or non-steroidal anti-inflammatory drugs (NSAIDs), history of alcoholism or current excessive alcohol intake, history of previous intentional overdose of paracetamol, previous suicide attempt or current unstable depression, body mass index <16 kg/m2, pregnant or breast-feeding women or women not using adequate contraception and participants unsuitable for BHR challenge testing in accordance with American Thoracic Society (ATS) criteria.33
Interventions
Participants were randomised to receive one of two treatment regimens for 12 weeks. The treatments were paracetamol 1 g, administered as two 500 mg tablets, or placebo administered as two identically appearing tablets, taken twice daily. The paracetamol and placebo tablets were supplied by Aspen Asia Pacific Ltd, Sydney, Australia. All participants were instructed to avoid taking other forms of paracetamol (including over-the-counter remedies containing paracetamol) or NSAIDs for the duration of the study. All participants were provided with a prescription for codeine to use as an analgesic during the study.
Randomisation
A computer-generated randomisation schedule was generated by the study statistician and was administered by the study pharmacists. It was necessary to randomise the participants prior to their final eligibility screening visit (visit 2) to enable the study pharmacists adequate time to prepare the study medication for dispensing at visit 2 following final determination of eligibility. If a participant failed one of the eligibility criteria at visit 2, the randomised medication was not dispensed and the participant was withdrawn from the study. The randomisation code was not reused.
Blinding
Study investigators, participants and participant healthcare providers were blinded through provision of medication as identically appearing tablets in bottles, with neither the investigator dispensing the medication nor the participants aware of the allocated treatment.
Design
The trial involved four study clinic visits and between two and four additional blood tests over 13 weeks (figure 1). A screening visit (visit 1) was held approximately 1 week prior to baseline and consisted of a medical history and brief physical examination, pregnancy test where applicable, bronchodilator reversibility testing, liver function screen and allergy skin prick tests (see online supplementary for details). A diary was used to record morning and evening peak expiratory flow (PEF) values (prior to asthma medication use) for 1 week prior to the second visit. Participants who met initial eligibility criteria were randomised at this stage, prior to final eligibility assessment at visit 2.
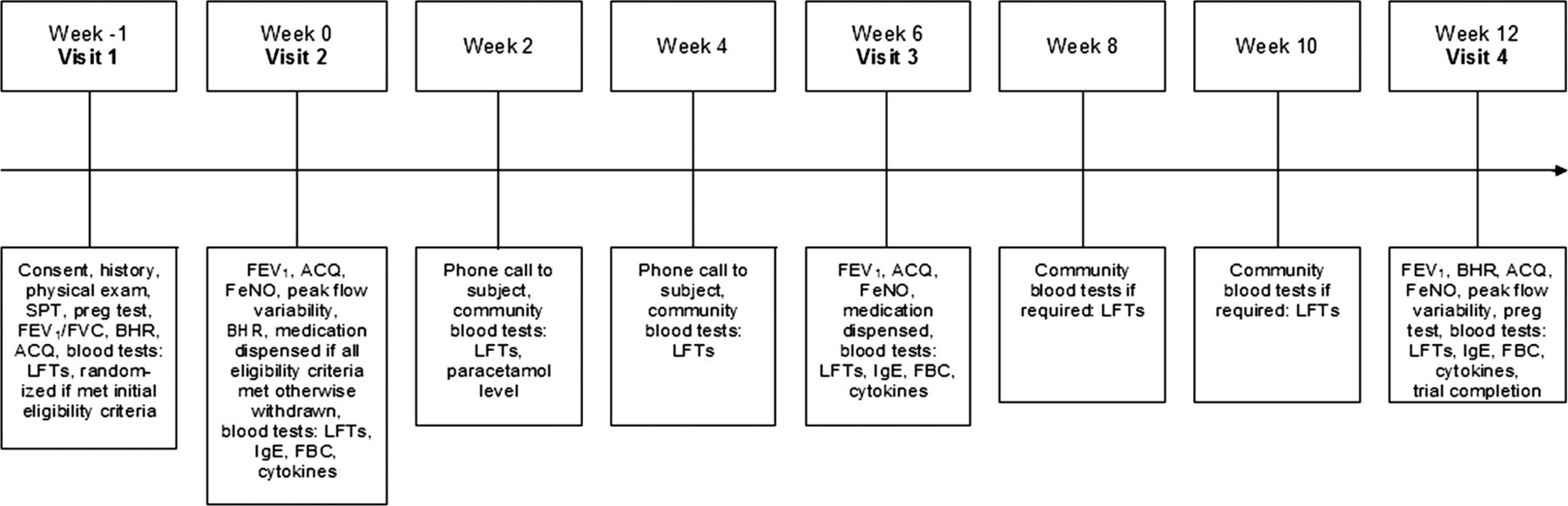
Study design flow chart. SPT, skin prick test; Preg test, pregnancy test; FEV1/FVC, forced expiratory volume in 1 s/forced vital capacity; BHR, bronchial hyper-responsiveness testing; ACQ, Asthma Control Questionnaire; LFT, liver function test; FeNO, fractional exhaled nitric oxide; IgE, immunoglobulin E; FBC, full blood count.
At visit 2, designated the baseline visit, the Qoltech asthma control questionnaire (ACQ)34 was administered and PEFvar (PEF variability measured as the amplitude as a percentage of the mean) calculated. Baseline assessments of FEV1 were undertaken using a micro medical microlab spirometer (Micro Medical, Kent, UK) and fractional exhaled nitric oxide (FeNO) was assessed using a NiOX Flex chemiluminescence analyser (Aerocrine AB, Stockholm, Sweden). Methacholine (Methapharm, Ontario, Canada) challenge testing was undertaken via the 2 min tidal breathing dosing protocol recommended by the ATS,33 as outlined in the online supplementary. Participants who met all the eligibility criteria were then dispensed a 6-week supply of randomised medication, a medication diary to record administered doses and a prescription for codeine phosphate for emergency pain relief during the trial period. These participants then underwent blood tests including full blood count (eosinophils), total serum immunoglobulin E (IgE) and serum cytokine levels (interferon (IFN)-γ, interleukin (IL)-4, IL-5, IL-13; see online supplementary for details).
At visits 3 and 4, 6 and 12 weeks after baseline, FEV1, ACQ, FeNO and blood tests were repeated and medication compliance checked via pill count and medication diary check (see online supplementary for details). At the third visit, participants were given a further 6-week supply of study medication, a second medication diary and a diary to record morning and evening PEF values in the final intervention week. At the fourth and final visit, BHR testing was repeated. Liver function tests were monitored throughout the study (see online supplementary for details).
Outcomes
The primary outcome variable was PC20 MCh at 12 weeks, adjusted for baseline. This direct measure of BHR was chosen as an objective well-standardised physiological measure of asthma severity, recommended for monitoring the effects of therapy which may modify asthma severity.33 ,35 Secondary outcome measures were FEV1, FEV1% predicted, ACQ score and FeNO at 6 and 12 weeks, and the mean morning peak flow, PEFvar, and exacerbations of asthma (requiring a doctor's visit and need for prednisone or nebulised bronchodilator) at 12 weeks. Blood eosinophil, serum IgE and serum cytokine (IFN-γ, IL-4, IL-5 and IL-13) levels were measured at 6 and 12 weeks.
Statistical methods
The primary analysis method was analysis of covariance (ANCOVA). The logarithm base two PC20 for methacholine at 12 weeks was the primary response variable, with the baseline logarithm base two PC20 as a covariate and a categorical variable for the paracetamol group. The difference in logarithm base two PC20 was the doubling dose difference between the two randomised groups. Secondary outcome variables, including FEV1, FEV1% predicted, ACQ score, FeNO, mean morning peak flow and PEFvar were also analysed by ANCOVA. The distribution of FeNO, serum IgE and eosinophil count was skewed and normality assumptions for these variables were best met on the natural logarithm scale.
The proportion of participants with at least one asthma exacerbation was compared as an absolute risk difference, with an appropriate CI, because in the event there were no asthma exacerbations in one of the randomised groups, and as a result a relative risk could not be calculated. Simple t tests were used to compare mean values for ALT by randomised group. FeNO, eosinophil count and IgE were logarithm transformed because of skewed distributions, and the difference in logarithms was compared by a t test. For those three variables with a logarithm transformation, the exponent of the difference in logarithms is interpreted as the ratio of mean values.
The analysis was by intention to treat randomised participants who passed the final eligibility screening and as a result received randomised treatment. Randomised participants who failed the final eligibility screen did not receive randomised treatment or undergo any outcome assessments. For each individual analysis, a two-sided p value of 0.05 was used, with 95% CIs for each estimate. We have not adjusted for multiple statistical testing.
Sample size
A sample size of 60 in each group has 80% power at the 5% level of significance to detect a difference of one doubling dose in PC20 MCh between the groups, based on an SD of 1.9.36 To allow for the possibility of up to 10% of study participants withdrawing early from the study, a recruitment target of 66 participants was set for each group.
Results
Recruitment started in June 2009 and ended in September 2011. The planned study period of 2 years was extended by 3 months due to difficulties in recruitment. Figure 2 shows the flow of participants. There were 724 patients assessed for eligibility by phone screening and/or at visit 1; of these, 338 failed to meet the inclusion criteria and 205 declined to participate (see online supplementary). There were 181 participants randomised prior to visit 2 based on initial eligibility at visit 1; 91 to the paracetamol group and 92 to the placebo group. Of the total number of participants allocated to the paracetamol group 53/91 were withdrawn, and 34/92 were withdrawn from the placebo group at visit 2 as they either did not meet the inclusion/exclusion criteria (PC20>16 mg/mL, n=68; PC20 <0.125 mg/mL, n=3; FEV1 <70% predicted, n=6; unable to perform spirometry, n=1) or were lost to follow-up or withdrew consent (n=9). Study medication was not dispensed to the participants who were withdrawn at visit 2 (see online supplementary).

{kind=link}
{kind=link}
CONSORT participant flow diagram.
Medication was dispensed to 94 participants who started the intervention phase following visit 2: 36 randomised to paracetamol and 58 to placebo. The characteristics of participants are shown in table 1. The mean age of participants was 40 years and there were 59 female participants. Approximately 30% of study participants were prescribed inhaled corticosteroids and 18% prescribed long-acting β agonist drugs. Around 90% of participants had positive skin prick tests to either cat, mixed grass or house dust mite. Participants had mild-to-moderate asthma, with a baseline ACQ score of 0.86 (SD 0.59). The baseline mean FeNO was 48.9 ppb (SD 41.3) and the mean FEV1 was 94% of predicted (SD 12.0). The baseline mean PC20 was 4.29 mg/mL (SD 4.54).
Characteristics of participants who received randomised treatment
There were 85/94 participants who completed the study. Five participants were withdrawn from the paracetamol group; two withdrew at the participant's own discretion, one was excluded due to a raised ALT (119 IU/L), one was lost to follow-up and one was excluded due to intercurrent illness. Four participants were withdrawn from the placebo group; two were excluded due to a raised ALT (207 and 227 IU/L, respectively), one withdrew at the participant's own discretion and one was lost to follow-up.
Primary outcome variable
At 12 weeks the mean (SD) logarithm base two PC20 was 1.07 (2.36) in the control group (N=54) and 0.62 (2.09) in the paracetamol group (N=31). After controlling for baseline PC20, the difference (expressed as a doubling dose difference, paracetamol minus placebo) was not statistically significant: −0.48 (95% CI −1.28 to 0.32), p=0.24 (table 2).
Effect of paracetamol use on BHR, lung function and asthma control
Secondary outcome variables
There were no statistically significant differences in FEV1, FEV1% predicted, ACQ score, mean morning peak flow or PEFvar between the control and paracetamol groups at week 12 (table 2), or in FEV1 or ACQ score at week 6 (see online supplementary). There were three asthma exacerbations in the placebo group and none in the paracetamol group, an absolute difference of 5.6% (95% CI −0.5% to 11.7%). There was 93.2% compliance in the control group and 90.8% compliance in the paracetamol group when assessed by pill count and medication diaries, a difference of 2.4% (95% CI −1.0% to 5.8%). Serum paracetamol levels (greater than the 30 μmol/L threshold) were detectable in between 31.3% to 38.7% of participants in the paracetamol group and were undetectable in all participants in the placebo group between weeks 2 and 12 of the study.
There were no statistically significant differences observed in log FeNO at week 6 (see online supplementary), at week 12, in log eosinophil or log IgE levels between the two groups at week 12 (table 3). Only a proportion of participants had measurable levels of IFN-γ, IL-4, IL-5 and IL-13 at baseline or at other times throughout the trial, precluding meaningful analysis (see online supplementary). ALT levels were significantly higher in the paracetamol group, with a mean ALT of 25.4 (SD 9.7) and 19.0 (SD 6.0) in the paracetamol and placebo groups, respectively, at visit 4, difference 6.3 (95% CI 2.9 to 9.7, p<0.001).
Effect of paracetamol use on FeNO, blood eosinophil count and serum IgE
Discussion
This double-blind, randomised, placebo-controlled, parallel group study found no statistically significant increase in BHR with 12 weeks of paracetamol treatment. However, the results did not rule out a clinically significant effect, with the 95% CI containing the prespecified difference of one doubling dose reduction in PC20. There were no significant differences observed in any of the prespecified secondary outcome variables of asthma control, inflammatory or immunological markers.
This is the first reported randomised placebo-controlled trial of the effects of the use of daily paracetamol in stable adult asthma. The only other published randomised controlled trial of paracetamol and asthma was the Boston University Fever Study.18 Children randomised to the ibuprofen group had a reduced risk of having an outpatient visit for asthma during the 4-week study period (OR 0.56, 95% CI 0.34 to 0.95) compared with children in the paracetamol group. Because the study did not include a placebo treatment, it was not possible to determine whether the observed difference in morbidity according to treatment group was attributable to an increased risk with paracetamol or a decreased risk with ibuprofen.
There are several methodological issues relevant to the interpretation of our study findings. First, as enshrined in the Declaration of Helsinki37 there is a requirement to study the least vulnerable populations wherever applicable. Most, but not all, of the putative adverse effects of paracetamol on asthma have been shown in observational studies of children and suggest that paracetamol may increase the risk of developing asthma.1 ,2 However, as there is some data to suggest that regular paracetamol use may lead to a deterioration in asthma control in adults,1 ,10 we opted to first examine the effects of paracetamol in adults with stable asthma.
Second, this trial was powered to determine whether there was an effect on BHR of at least one doubling dose reduction in PC20 MCh. Our ability to achieve the designated sample size completing the study was affected by several factors. First, despite a rigorous recruitment campaign during which over 700 patients were screened, due to the inclusion and exclusion criteria employed to ensure participant safety, only 94 screened participants were dispensed randomised medication. Second, variability in PC20 from baseline to week 12 was larger than anticipated, with a pooled SD of 2.27 doubling doses compared with that used in the sample size calculation based on an SD of 1.9, derived from previous studies.36 Another factor that affected the study power was the requirement to randomise participants prior to their final screening visit in order to allow the pharmacy adequate time for dispensing at visit 2, following final determination of eligibility. If the participant failed this final eligibility, the randomised medication was not dispensed, the participant was withdrawn from the study and the randomisation code was not reused. By chance, this resulted in a disparity between the proportion of participants receiving active and placebo study medication. The power was reduced further due to the withdrawal of 10% of participants after randomised treatment was dispensed. As there is an uncertain association between observed variables and missing BHR data in these participants, it was not possible to perform a robust imputation.
Compliance was high when measured via pill count, and although less than half of participants in the paracetamol group had measurable levels of paracetamol in the blood at the times tested throughout the study, this is likely to be due to the laboratory cut-off for a detectable paracetamol level (30 μmol/L). Following a 1 g dose, participant blood levels may fall below this laboratory cut-off level in as little as 3 h (given a paracetamol half-life of 2 h and a peak plasma concentration 1 h after administration of 80 μmol/L38). The use of this laboratory cut-off for paracetamol levels meant that it was not possible to investigate medication compliance through this method.
Our 12-week dosing period was chosen based on evidence that regular, long-term use of paracetamol is associated with an increased risk of asthma in adults9–11 ,16 ,17 and that chronic ingestion of therapeutic doses can reduce serum antioxidant capacity in as little as 2 weeks.39 We had originally intended to use the maximum daily dose of 4 g paracetamol, however, chose to administer half this dose due to concerns of liver toxicity. These concerns were based on a previous clinical trial of paracetamol in which the incidence of ALT elevations more than three times the upper limit of normal in healthy participants taking 4 g/day for 14 days was 31–44%.40 Our results showed no clinically significant liver function derangement with paracetamol administered at a dose of 2 g/day for 12 weeks.
While the study did not demonstrate a statistically significant effect of paracetamol on BHR to MCh, the results do not rule out a clinically significant effect, with the upper 95% CI of a 1.28 doubling dose worsening in BHR containing the prespecified difference of one doubling dose. Furthermore, our point estimate of a reduction in PC20 of 0.48 of a doubling dose could potentially be of major public health significance. As proposed by Mitchell,41 a small shift to the left of the BHR curve in a population could lead to a relatively large increase in the prevalence of severe asthma. Relevant to the interpretation of our findings it has recently been calculated that a one half doubling dose increase in BHR increases the prevalence of moderate and severe BHR by about 30%.42 Likewise, although the 9% increase in FeNO with paracetamol was not statistically significant, a change of this magnitude is considered clinically significant.43 For FEV1, the point estimate was consistent with a lower value in the paracetamol group, however, the difference was of uncertain clinical significance and was associated with wide CIs.
No significant effect was seen on serum IgE or peripheral blood eosinophil levels. It was not possible to undertake any meaningful analysis of the cytokine measurements due to the low numbers of participants with detectable levels, and as a result we were unable to determine whether paracetamol influenced the Th1/Th2 balance. Another less recognised potential mechanism of action, which was not directly assessed in this study, relates to neurogenic inflammation of the airways through the stimulation of the transient receptor potential ankyrin-1 cation channel by N-acetyl-p-benzoquinoneimine, the metabolite of paracetamol.26 This pathway, which is activated following therapeutic doses of paracetamol, mediates a non-eosinophilic inflammatory response and has been implicated in the pathogenesis or provocation of asthma by isocyanates, aldehydes, cigarette smoke and chlorine.44 ,45
Our findings provide information on which the design of further studies could be based. A trial of similar design, utilising the same duration and dose of paracetamol and with BHR testing to MCh as the primary outcome variable, based on the SD derived from this study, would require a sample size of approximately 650 to attain adequate power to detect a difference of 0.5 doubling doses. Alternatively, a study of short-term use of paracetamol at higher doses could be undertaken, to more closely replicate the common use of paracetamol for relief of fever or pain in self-limited illnesses. Based on our findings, a sample size of 140 would be adequate to determine a 0.5 doubling dose difference in MCh BHR, and a 10% increase in FeNO, in a short-term study of crossover design. Important issues with the design of such a study are the duration of the treatment periods and the crossover period. It would be important if possible to include a placebo rather than ibuprofen arm, as NSAIDs may have the potential to both cause NSAID-induced bronchospasm, as well as reducing asthma severity with long-term use.29
Finally, our study investigated the effect of paracetamol on asthma severity and not whether paracetamol has a role in the pathogenesis of asthma. Testing this hypothesis would require clinical trials of the effect of paracetamol use in pregnancy on the development of asthma in childhood and the effect of paracetamol use in infants and older children and subsequent asthma risk. Such studies would raise ethical and practical issues regarding consent and the use of placebo for the management of pain or fever during pregnancy and in young children. However, given the common usage of paracetamol in all age groups including pregnancy and the global burden of asthma, we propose that randomised controlled trials are required to determine the effect of paracetamol use on the development of asthma in infancy and early childhood.
In conclusion, this study has shown no significant effect of 12 weeks of treatment with paracetamol at half the maximum therapeutic daily dose on BHR and asthma control in adults with well-controlled asthma. While this outcome provides some reassurance that regular paracetamol use has no marked deleterious effect in adult asthma, further adequately powered studies are needed before the safety of paracetamol for patients with asthma is assured. Furthermore, the study findings do not preclude an effect of paracetamol on the development of asthma in infancy, childhood or adult life.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors SJI and RB contributed to study planning, study conduct, data analysis and manuscript preparation. KP contributed to study planning, study conduct and manuscript preparation. MW, SJ and MP contributed to study conduct. MW contributed to randomisation, statistical analysis and preparation of manuscript. RS contributed to blood analysis and preparation of manuscript. JC contributed to study planning and manuscript preparation. JT and PS were safety investigators.
-
Funding Funding for the study was through research grants from the Health Research Council of New Zealand, the Wellington Medical Research Foundation, the Asthma and Respiratory Foundation of New Zealand and the University of Otago.
-
Competing interests RB has been a member of the GlaxoSmithKline (NZ) Advisory Board, and received research grants, payment for lectures or support to attend meetings from GlaxoSmithKline, a manufacturer of paracetamol. SJI is a Health Research Council Clinical Research Training Fellow.
-
Ethics approval The study was approved by the Central Regional Ethics Committee (CEN/08/12/070) and all participants gave written informed consent. The trial was registered on the Australian New Zealand Clinical Trials Registry (ACTRN12609000551291).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement No additional data are available.