Article Text

Abstract
Introduction Sideroblastic cardiomyopathy secondary to repeated blood transfusions is a feared complication in thalassaemia. Control of myocardial iron is thus becoming the cornerstone of thalassaemia management. Recent evidence suggests a role for L-type Ca2+ channels in mediating iron uptake by the heart. Blocking the cellular iron uptake through these channels may add to the benefit of therapy to standard chelation in reducing myocardial iron. We aim to determine the efficacy of amlodipine (a calcium channel blocker) as an adjunct to standard aggressive chelation in retarding myocardial iron deposition in thalassaemics with or without cardiomyopathy.
Outcomes The primary outcome is to compare the efficacy of amlodipine+chelation (intervention) versus standard chelation (control) in retarding myocardial iron deposition. Secondary outcomes include the effect of amlodipine therapy on systolic and diastolic function, strain and strain rate and liver iron content.
Methods and analysis This is a single-centre, parallel-group, prospective randomised control trial. Twenty patients will be randomised in a 1:1 allocation ratio into the intervention and control arms. In addition to conventional echocardiography, MRI T2* values for assessment of cardiac and liver iron load will be obtained at baseline and at 6 and 12 months. Cardiac T2* will be reported as the geometric mean and per cent coefficient of variation, and an increase in cardiac T2* values from baseline will be used as an end point to compare the efficacy of therapy. A p Value of <0.05 will be considered significant.
Study setting Department of Pediatric and Child Health, Aga Khan University Hospital, Karachi, Pakistan.
Ethics and dissemination This study has been approved by the Ethics Review Committee and Clinical Trials Unit at The Aga Khan University with respect to scientific content and compliance with applicable research and human subjects regulations. Findings will be reported through scientific publications and research conferences and project summary papers for participants.
Trial registration number ClinicalTrials.Gov. Registration no: NCT02065492.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
Amlodipine for Myocardial Iron in Thalassemia (AMIT) is the second randomised control trial to date aimed at assessing the role of amlodipine in reducing moderate-to-severe myocardial iron load secondary to repeated blood transfusions.
-
Our study will also be the first to assess the association of changes in cardiac T2* values with important clinical parameters of cardiac function such as echocardiographic estimates of left ventricular ejection fraction (LVEF), systolic and diastolic function as well as myocardial strain and strain rate analysis, at 6 and 12 months following amlodipine therapy.
-
Owing to budget restrictions, we will not be able to achieve the calculated sample size. Our pilot study has helped establish the T2* modality in Pakistan and will also help us determine the feasibility to perform a larger trial with the goal of recruiting the calculated sample size.
-
Similarly, we will not be looking at the responses of amlodipine controlling for the type of iron chelator or regimen used.
Introduction
Thalassaemia, a hereditary anaemia of varying severity, is one of the most common inherited disorders in Pakistan.1 Approximately 9000 children with β thalassaemia are born every year, although no thalassaemia registry is available in Pakistan. The estimated carrier rate is 5–7%, which amounts to 9.8 million carriers in the entire population.2
Haemosiderosis due to transfusion treatments is a major cause of death in patients with thalassaemia major.3 Iron overload can lead to iron deposits in many tissues, particularly in the heart. It has also been shown to be associated with elevated oxidative stress in tissues. Iron overload conditions can lead to increased iron uptake into myocardial cells, resulting in myocardial damage and failure.4–7 Catastrophic deterioration in cardiac function resulting in death may occur rapidly once clinically obvious heart failure is present.8 Therefore, heart disease is the most important complication and the major determinant of survival in patients with thalassaemia, responsible for more than half of the deaths in this population.9 ,10 It may take the form of cardiomyopathy, pulmonary hypertension, heart failure, arrhythmias, pericarditis and myocarditis.9–12
Currently, the mechanism of iron uptake into myocardial cells is not clearly understood. Growing evidence suggests that L-type Ca2+ channels (L-TCCs) are a possible pathway for ferrous iron (Fe2+) uptake into myocardial cells under iron overload conditions.13 Several findings have been shown to support the role of L-TCC in myocardial iron transport.14 ,15 A study in an iron loaded rat heart showed that iron uptake was increased by the L-TCC agonist Bay K 8644 and inhibited by the L-TCC blocker nifedipine.14–16 Oudit et al16 demonstrated that treatments with L-TCC blockers such as amlodipine and verapamil could not only lead to the inhibition of the L-TCC current in myocardial cells, but also reduced myocardial iron accumulation, decreased oxidative stress and improved survival in iron-loaded mice.
Since iron overload patients can develop cardiomyopathy and heart failure,11 ,17 ,18 which serves as the major cause of death in these patients, it is important that the use of drugs such as amlodipine be studied for the prevention of myocardial iron deposition.
Current clinical practice
In thalassaemia major, the current practice to prevent iron overload is to start chelation therapy after the first 10–20 transfusions or when serum ferritin levels rise above 1000 µg/L. The standard recommended method of administration of chelation therapy is slow subcutaneous infusion over 8–12 h of 10% deferoxamine (desferal) solution (DFO), using an infusion pump or oral deferasirox (DFX) or oral deferiprone (DFP). The standard dose of deferoxamine is 20–40 mg/kg for children and up to 50–60 mg/kg for adults as an 8–12 h subcutaneous infusion for a minimum of 3–5 nights a week. Patients with high degrees of iron loading or those at increased risk of cardiac complications require adjustments in chelation therapy. The dose of oral deferasirox is 20–40 mg/kg/day. Recent studies suggest a possible synergistic effect of combination therapy of DFP with DFO, especially in reducing the myocardial iron load. This has led to multiple studies aimed at assessing the effectiveness of DFO, DFP and DFX either as monotherapy or as part of combination therapy in patients with transfusion-dependent thalassaemia. The multicentre, prospective Evaluation of Patients’ Iron Chelation with Exjade (EPIC) trial has demonstrated the effectiveness of DFX as monotherapy in reducing mild-to-moderate and severe myocardial iron load, as documented by improvements in cardiac T2* values at 12 months,19 along with persistence of these improvements from baseline even after 24 and 36 months of deferasirox treatment when evaluated in a substudy of these patients.20 ,21 Pepe et al22 demonstrated that the combined DFP+DFO regimen and DFP in monotherapy were not significantly different in removing myocardial iron and improving heart function, when followed up to 18 months. Similarly, in a meta-analysis by Maggio et al,23 myocardial iron concentration measured as T2* was found to be not statistically significantly different in the DFP+DFO versus the DFO-treated groups. This is in contrast to findings by Tanner et al,24 who have demonstrated significant improvements in the combined treatment group compared with the deferoxamine group in myocardial T2* as well as absolute LVEF. Thus, algorithms involving single or combination chelation therapies have been developed and are used in different centres. As documented by Veríssimo et al,25 the Scientific Committee of Associacao Brasileira de Thalassemia (ABRASTA) have proposed an evidence-based iron chelation protocol for patients with thalassaemia under regular transfusions, directed by the myocardial T2* values obtained. Attached as online supplementary appendix A, the protocol proves useful in helping to initiate appropriate chelation therapy particularly in the setting of severe myocardial iron overload.
With the growing evidence that suggests L-TCCs as a possible pathway for ferrous ions (Fe2+) uptake into myocardial cells,13 we hypothesise that the use of L-type calcium channel blockers such as amlodipine will lead to a reduction in this myocardial iron deposition.
Null hypothesis
There is no difference between the efficacy of chelation plus amlodipine therapy and chelation therapy alone in retarding the rate of myocardial iron deposition in patients with thalassaemia with iron overload and a constant transfusion need.
Alternate hypothesis
Chelation plus amlodipine therapy is more efficacious than chelation therapy alone in retarding the rate of myocardial iron deposition in patients with thalassaemia with iron overload and a constant transfusion need.
Primary objective
The aim of our study is to determine if amlodipine, an L-type specific calcium channel blocker, in addition to the standard aggressive chelation therapy, can retard the deposition of iron in the myocardium of patients with thalassaemia with significant myocardial iron load with or without cardiomyopathy.
Secondary objectives
To determine if there is a difference in LV size, systolic and diastolic function in patients receiving amlodipine plus chelation therapy when compared with patients who receive only chelation therapy.
Methods
Operational definitions
-
Myocardial iron load (see online supplementary appendix B for study protocol):
-
T2*: The myocardial T2* values in the mid-ventricular septum in the normal participants using a multiechocardiography acquisition is equal to are 33.3±7.8 ms.26 A shortening of myocardial T2* to <20 ms (implying increased myocardial iron) is associated with an increased chance of decreased LV function.27
-
Severity index based on T2*:25
-
Normal T2* >20 ms,
-
Mild 15–20 ms,
-
Moderate 10–15 ms,
-
Severe <10 ms.
-
-
Truncation model: Short T2* values (4–10 ms) lead to a rapid decay in signal intensity with the signals of later echocardiography images buried in the background noise and motion. Therefore, in order to make the best-fit curve to an monoexponential curve, all data points less than 2 SNR (signal-to-noise ratio) will be removed.
-
-
Liver Iron load: The values for T2* in normal participants for the liver with standard acquisition and multiechocardiography acquisition will be taken as 26.6±4.7 ms and 26.7±4.2 ms, respectively.26
-
LV systolic dysfunction: LV systolic dysfunction will be defined as following:
-
EF z-score >−2: normal
-
≤−2 and >−3: mild
-
≤−3 and >−4: moderate
-
≤−4: severe.
-
-
Diastolic dysfunction: Diastolic dysfunction is defined by echocardiograph through stages 1–4, using mitral inflow, pulmonary venous flow and Doppler tissue imaging of mitral annular motion as described by Khouri et al.28 Mitral inflow at peak Valsalva and colour M mode propagation velocity will not be done.
-
Congenital or acquired heart disease: Significant congenital defects will include cyanotic heart diseases, acyanotic heart diseases (atrial septal defect, ventricular septal defect, patent ductus arteriosus), with moderate-to-severe left to right shunt (as determined by the treating paediatric cardiologist), and moderate-to-severe valvular disease (mitral stenosis/mitral regurgitation, aortic stenosis/aortic regurgitation, tricuspid regurgitation/tricuspid stenosis, pulmonary stenosis/pulmonary regurgitation) whether congenital or acquired (other than thalassaemia).
-
Heart failure: Defined as patients with known signs and symptoms of heart failure, such as worsening dyspnoea at rest or during exercise or exercise intolerance (NYHA functional II and above).
Study design
The use of amlodipine to treat hypertension has been established clinically. The role of this medication in retarding myocardial iron uptake in increased iron states has been demonstrated in mice models. To date, only a single study has been published to assess the role of amlodipine as an iron uptake inhibitor.29 Our study will therefore be the largest study to date, and is a hybrid phase 2 and 3 randomised controlled trial. Utilising a prospective randomised controlled method, patients presenting to a single-center will undergo parallel group, simple randomisation with a 1:1 allocation.
Sample size calculation
To date, there is only a single human study that has been reported, and therefore limited information is available for sample size calculation.
With the information gathered from Fernandes et al,29 using PASS 11 software, using a one-sided two-sample t test, we calculated that group sample sizes of 23 in each arm (intervention and control) achieve 90% power to detect a difference of −6.6 between the null hypothesis that both group means are 21.7 and the alternative hypothesis that the mean of group 2 is 28.3 with known group SDs of 7.2 and 8.0 and with a significance level (α) of 0.05000.
However, the sample size for our current study is dictated by the grant budget. Our sample will therefore be 20 study participants. Given the 1:1 allocation ratio, 10 participants each will be randomised into either one of two groups; the control group (arm A) will comprise of standard chelation therapy alone while the intervention group (arm B) will receive amlodipine in addition to the standard chelation. Our future work and grant application will encompass larger studies to recruit more patients divided into different subgroups of chelation.
Treatment arms
-
Arm A: Receiving chelation therapy alone
-
Arm B: Receiving chelation therapy plus amlodipine.
Study population
Paediatric patients with thalassaemia aged 6–20 years managed in the outpatient haematology clinics at Aga Khan University Hospital (AKUH) will be considered for enrolment if they meet the inclusion criteria.
Eligibility criteria
The following inclusion/exclusion criteria will be used for selection:
Inclusion criteria:
-
Paediatric patients aged ≥6 and ≤20 years managed at AKUH for at least 1 year;
-
≥10 blood transfusions in a lifetime;
-
Transfusion need ≥180 mL/kg/year;
-
Serum ferritin ≥1000 µg/dL;
-
Patient deemed capable of receiving chelation therapy (by treating haematologist) either by subcutaneous infusion of deferoxamine (desferal; 3–5 days a week) or oral deferasirox (daily) or deferiprone (oral) or a combination of desferal and deferiprone;
-
Patients who have been on a stable chelation regimen ≥6 months;
-
Completed and signed informed consent/assent.
Exclusion criteria:
-
Patients with known hypersensitivity to amlodipine.
-
Patients with known sinoatrial nodal disease or aortic stenosis.
-
Patients with known severe myocardial dysfunction, defined as an LVEF of ≤4 SD for age even without symptoms.
-
Patients with known signs and symptoms of heart failure.
-
Patients with a T2* value of <4 ms on cardiac MRI.
-
Patients with systolic blood pressure (SBP) ≤2 SD for age (systemic hypotension) at the time of enrolment.
-
Patients with previously diagnosed significant congenital heart diseases or acquired heart diseases other than thalassaemia (as defined earlier).
-
Patients with known contraindications to MRI (pacemakers, cerebral aneurysm metal clips, etc).
-
Patient with a known history of developing tetany after use of a calcium channel blocker.
-
Known pregnancy.
Patient recruitment and randomisation process
As shown in online supplementary appendix B, once eligible, parents of the patients presenting to the haematology outpatient clinics will be approached by the principal investigator (PI)/co-investigator for administering informed consent and assent, respectively, and as applicable on the day of admission. As per Good Clinical Practice (GCP) guidelines (GCP 4.8.11), a copy of the signed dated informed consent will be provided to the patient and the patients’ legally acceptable representative.
After obtaining informed consent, the patients will be recruited in the trial. The patients will be randomly assigned to either of the two intervention arms, amlodipine (0.1 mg/kg/day or maximum of 2.5 mg/day) plus chelation therapy or chelation therapy alone. Amlodipine is administered as a single daily dose. Recruited patients will receive treatment accordingly. Randomisation will be computerised; allocation concealment will be exercised by sequential participant identification, and arms randomised using sealed opaque envelopes.
As per established clinical guidelines in Pakistan, as seen in online supplementary appendices C and D of the protocol, owing to the unavailability of cardiac MRI T2* technology, the chelation dosing and types used have commonly been dictated by serum ferritin levels.
Having recently acquired the technology of cardiac T2* MRI for this project, the modality and its clinical use has been now introduced in Pakistan, and we are now informally basing the chelation on the values of T2* obtained, as per the guidelines suggested by Veríssimo et al (see online supplementary appendix A) until more updated local guidelines are available at our institution. The chelation therapy is therefore left to the discretion of the treating haematologist. Since it is a single centre study and the patients are predominantly managed by two haematologists, we have been able to initiate the informal chelation protocol in our study patients, thus introducing standardisation of care. This, along with our current randomisation process, we hope, will help equally distribute the effect of the chelation in both the arms. On the basis of this reason, we have decided not to randomise on the type of chelation therapy.
Blinding
While the primary physician and the patients will be aware of the allocated arm, the readers involved in T2* assessment will be blinded to the allocated arm. Similarly, outcome assessors and data analysts will also be blinded to the allocation.
Additional measures to ensure data safety and monitoring along with validation will be made, the details of which will be discussed later in this protocol.
Study drug: amlodipine
Amlodipine is a long-acting L-type calcium channel blocker that needs to be taken only once a day without any dietary restrictions. Numerous studies have shown that it is better tolerated when compared with other L-type calcium channel blockers like nifedipine.30 It is also available at an easily affordable price that allows maximum compliance and makes the drug highly suitable for our study. Based on the studies available to date, amlodipine should be initiated in children at a dose of 0.05–0.1 mg/kg/day given once daily. Younger children appear to require higher doses, often as much as 0.3–0.4 mg/kg/day. The adult dose is 2.5–5 mg/day.31
The symptomatic adverse reactions produced by amlodipine are tolerable and, if they become severe, can be treated symptomatically; these include abdominal pain, gastrointestinal (GI) disturbance, blurred vision, headache, flushing, palpitations, dizziness, somnolence, hypotension and cough. The severe adverse effects of amlodipine, which may give rise to further complications, include peripheral oedema and sinus bradycardia. The contraindications to amlodipine use are hypotension, aortic stenosis and sinoatrial node disease. The paediatric cardiology and haematology team will deal with adverse events (AEs) associated with amlodipine and treatment cost will be covered by the grant budget. The frequency of serious AEs (SAEs) requiring intervention frequency of SAEs requiring intervention is <1%. On this basis, we anticipate at the most one patient who may develop severe AEs requiring interventions like stopping the medication or adding a diuretic.
As part of the protocol, regular monthly follow-ups of all the study participants in our paediatric cardiology clinics will help us to ensure adequate compliance to the study drug. Moreover, the treating haematologist will also ensure compliance to the chelation therapy.
Chelation drug
Potential adverse effects
Deferoxamine
Deferoxamine may cause injection site reactions, systemic reactions (eg, abdominal pain, arthralgia, nausea, vomiting), cardiovascular reactions (eg, hypotension with too rapid IV infusion, tachycardia, shock), hypersensitivity reactions (eg, anaphylactic reaction with or without shock, angio-oedema, generalised rash, urticaria), haematological reactions (eg, thrombocytopaenia and leucopaenia), hepatic dysfunction, musculoskeletal reactions (eg, muscle spasms, growth retardation and bone changes including metaphyseal dysplasia are common in doses ≥60 mg/kg, especially those who begin iron chelation in the first 3 years of life; reduced risk if doses are kept to ≤40 mg/kg) and neurological disturbances (eg, dizziness, peripheral sensory, motor, or mixed neuropathy, paraesthesias, seizures; exacerbation or precipitation of aluminium-related dialysis encephalopathy), and respiratory reactions (eg, acute respiratory distress syndrome with dyspnoea, cyanosis and/or interstitial infiltrates) (chelation therapy protocol as per online supplementary appendix A or C and D).
Deferasirox
Deferasirox does not appear to present any increased overall burden in adverse effects when compared with placebo (chelation therapy protocol as per online supplementary appendix A or C and D). When compared with deferoxamine, there is an absence of injection site effects, but an increase in GI effects, which occurs in about 15% of patients. Other common effects include transient rises in serum creatinine, back pain and rash.
The haematology team as per their standard clinical practice will deal with adverse effects and the cost incurred will also be borne by the patient.
Study procedures and monitoring
Eligible patients meeting the inclusion/exclusion criteria will be recruited over a 4-month period (following registration on clinicaltrials.gov). Each patient will be randomised into either of two study arms: amlodipine plus chelation or chelation alone. All patients will undergo the following tests at baseline and at 6 and 12 month follow-up visits:
-
Myocardial iron content using cardiac MRI and T2* imaging;27 ,32
-
Myocardial systolic function using standard measures (EF) and diastolic function using conventional and tissue Doppler imaging;
-
Myocardial function using speckled tracking derived strain and strain rate analysis;
-
Serum ferritin levels.
The cost of the aforementioned investigations is budgeted through the grant except for the cost of the serum ferritin levels which will be borne by the patient. In addition, patients will receive standard care and have follow-up appointments as dictated by the chelation therapy. If found to be hypotensive (SBP <−2SD for age) or experiencing bradycardia (<−2SD for age adjusted heart rate), they will be evaluated by a paediatric cardiologist and managed accordingly. Patients who are persistently hypotensive and experiencing bradycardia will be taken off the medication and receive chelation only. These patients will be considered as part of the amlodipine arm during analysis (intension to treat). Dropouts or patients lost to follow-up during the first 6 months of being in the study will be replaced. Dropouts after completion of the 6-month evaluation will not be replaced.
The treating haematologist will determine chelation therapy modification based on myocardial iron content.
Adverse events—identification, reporting and clinical management
Definitions
Adverse event: An ‘Adverse Event’ (AE) is “any untoward occurrence (physical, psychological, social, or economic) in a human subject participating in research.” The event is undesirable and has an unintended outcome, but is not necessarily unexpected. The event may have been described in the informed consent as a risk of the study. AEs include abnormal laboratory findings, a symptom or disease temporarily associated with the use of an investigational agent, or the progression of disease, whether or not related to the medicinal (investigational) product (ICH GCP 1.2).
Serious adverse event: An ‘Serious Adverse Event’ (SAE) is any untoward medical occurrence that at any dose:
-
Results in death,
-
Is life-threatening,
-
Requires inpatient hospitalisation or prolongation of existing hospitalisation,
-
Results in persistent or significant disability/incapacity,
-
Is a congenital anomaly/birth defect (ICH GCP 1.50).
Identifying, recording and reporting SAEs:
-
Fatal
Any death occurring within the trial period or within 28 days after the last dose of the investigational product except for any death that is unequivocally due to progression of disease.
-
Life-threatening
The term ‘life-threatening’ in the definition of ‘serious’ refers to an event in which the patient was at risk of death at the time of the event; it does not refer to an event that hypothetically might have caused death if it were more severe.
-
Hospitalisation
If the AE resulted in admission to a hospital overnight, or necessitated prolongation of a stay in hospital. This excludes hospital stays for elective surgery or planned procedures.
-
Disabling or incapacitating
If the AE resulted in significant or persistent change, impairment, damage or disruption in the patient's ability to conduct normal life functions.
-
Congenital anomaly
If exposure by either parent to trial drug, before conception, or by the mother during pregnancy, is associated with the presence of developmental abnormalities at birth (ICH E2A).
All clinical adverse experiences and symptoms will be recorded by the clinical staff on ‘Adverse Event Report Forms’. These forms will be filled at the following time points:
-
At the 6-month study visit.
-
At the 12-month study visit.
-
At all visits to the Clinical Trials Unit (CTU) pharmacy for dispensing amlodipine. These are not study visits.
-
At all routine visits to the outpatient haematology clinic. These are not study visits.
-
At all monthly cardiology clinic visits that are part of the study.
The start date, severity, treatment or action taken, stop date or determination of ongoing status, and assessment of causality will be noted for each event. SAEs will be documented when identified and relevant follow-up information recorded when available.
Reporting AEs
All SAEs will be reported to the sponsor within five working days of the SAE being identified even if the event is considered to be not related to the investigational product. SAEs will be reported to the Institutional Review Board (IRB)/Ethics Committee (EC) per Aga Khan University's Institutional policy. Copies of reports of all SAEs will be kept at the study site. A summary of AEs will be submitted to the sponsor and the IRB/ECs at least annually and to other entities (eg, safety monitoring board) if applicable. The anonymity of the participant shall be respected when forwarding all information. The participant's name will not appear on any form or attachment.
Managing AEs
AEs that require symptomatic management only will be treated by the participant's primary haematologist. AEs that require hospitalisation will also be managed by the participant's primary haematologist and the costs incurred will be covered by the research fund. Cardiovascular AEs that require outpatient or inpatient management will be treated by the PI and his cardiology team and all costs incurred will be covered by the research fund.
Magnetic resonance and T2* imaging
Myocardial T2* MRI is the most sensitive and easily reproducible index of myocardial iron deposition currently available.32 Myocardial iron deposition can be reproducibly quantified using T2*. This is the most significant variable for predicting a requirement for targeted treatment of myocardial iron overload and it cannot be replaced by serum ferritin, liver iron or any other measurement. A shortening of myocardial T2* to <20 ms (implying increased myocardial iron) is associated with an increased chance of decreased LV function.27 For example, patients with T2* values >20 ms have a very low chance of decreased LVEF. T2* values of 10–20 ms indicate up to a 10% chance of decreased LVEF; 8–10 ms indicates an 18% chance; 6 ms indicates a 38% chance; and T2* values of just 4 ms indicate a 70% chance of decreased LVEF.33
The following index will be used to categorise myocardial iron deposition severity based on T2* values:25
-
Normal: T2* ≥20 ms;
-
Mild: T2* 15–20 ms;
-
Moderate: T2* 10–15 ms;
-
Severe: T2* <10 ms.
Excellent T2* reproducibility between scanners produced by two different manufacturers supports the feasibility of widespread implementation of the technique.32 ,33 Thus, the inclusion of cardiac T2* assessment, in concert with conventional long-term assessments of tissue iron loading, is mandatory for the comprehensive evaluation of iron loading.
Patients will be scanned using the single breath-hold multiecho technique. The multiecho sequence parameters will be as follows: for the measurement of myocardial T2*, a single short axis mid-ventricular will be acquired at 10 echo times (1.99–20.26 ms, which will increase in 2.03 ms increments) in a single breath-hold. A gradient-echo sequence will be used with a flip angle of 20°, a matrix of 90–256 pixels, a field of view of 40 cm, and a sampling bandwidth of 814 Hz per pixel. The TR in between the 10 radio frequency pulses applied to each cardiac cycle will be 22.17 ms. A homogeneous full-thickness region of interest (ROI) will be chosen in the LV septum, encompassing both epicardial and endocardial regions. The signal intensity of this region will be measured for each image using QMass V.7.6, MEDIS software, and will then be plotted against the echo time to form an exponential decay curve. To derive T2*, an exponential trend line will be fitted with an equation in the form y Ke–TE/T2* where K represents a constant, TE represents the echo time and y represents the image signal intensity.
Given the known risk of sideroblastic cardiomyopathy in the hearts of patients with thalassaemia, we expect our T2* values to reflect this myocardial iron load as substantially shorter (4–10 ms) values. These values can potentially lead to a rapid decay in signal intensity with the signals of later echocardiography images buried in the background noise and motion. Therefore, in order to make the best-fit curve, we will employ the truncation method as described in the literature,34 which will exclude all data points less than a particular SNR, generated according to the algorithm described by Bonny et al.35 For all practical purposes, as utilised in the literature, we will therefore remove data points <2 SNR in our study for short T2* values. Figure 1 demonstrates the practical application of the truncation model to account for background noise.

Adapted from Carpenter et al. This graph shows signal intensity (arbitrary units) plotted against echocardiography time (milliseconds) for the hearts shown on top. Heart 1 (with normal iron levels) has a shallow decay curve with a T2* value of >20 ms. Heart 2, owing to severe iron loading, has a much more rapid decay with T2* <10 ms.
Carpenter et al36 also attempted to provide calibration in humans for cardiovascular magnetic resonance relaxation parameter R2* (reciprocal of clinically measured T2*) against actual myocardial iron concentration. They reported that myocardial R2* provides a robust curvilinear relationship when calibrated against chemically assayed values of cardiac iron in postmortem studies. They also reported that R2* mid-ventricular septal ROI was highly representative of mean global myocardial iron (figure 2).
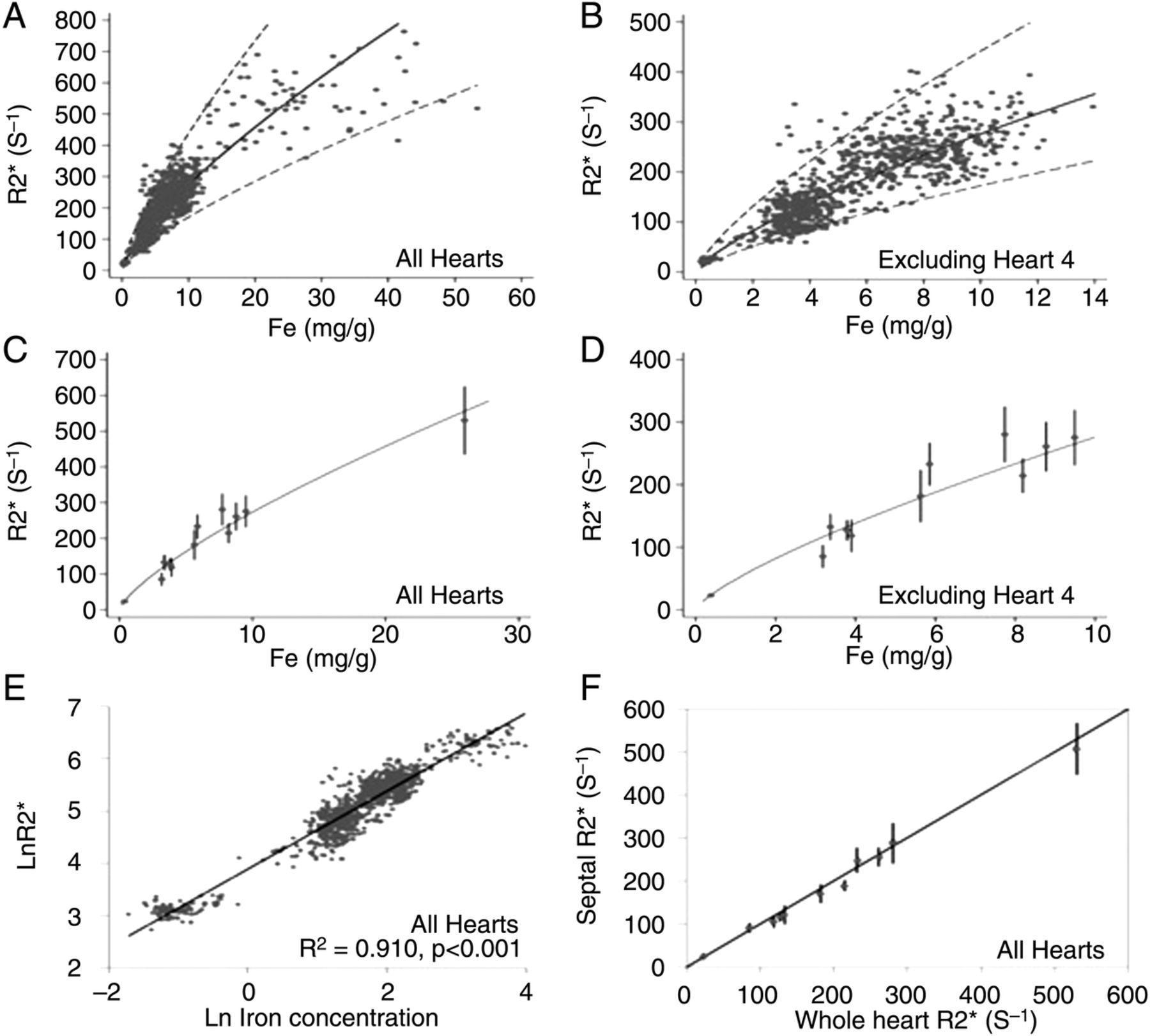
Adapted from Carpenter et al correlated T2* values to myocardial iron content. The regression (solid line) and 95% confidence bands (dotted lines) are shown and derived from analysis of the log-log data shown in E. (A) R2* plotted versus myocardial iron concentration measured from each myocardial region of interest (ROI). (B) R2* versus myocardial iron concentration excluding heart 4. (C) Mean R2* plotted versus mean iron concentration for each heart. (D) Mean R2* versus mean iron concentration excluding heart 4. (E) ln(R2*) plotted versus ln((Fe)) for all ROIs, including heart 4, showing the best-fit linear regression line. (F) Mean midseptal R2* versus mean whole-heart R2* plotted against the line of identity showing that the septal R2* value is highly representative of the whole-heart R2*.
Liver: The liver T2* value will be determined as follows: a single transaxial 10 mm slice through the centre of the liver will be scanned at a series of 14 different echo times (0.94–17.58 ms, which will increase in 1.28 ms increments), using a multiecho gradient-echo sequence with a flip angle of 20°, a matrix of 128–128 pixels, a field of view of 40 cm and a sampling bandwidth of 1502 Hz per pixel. Both even and odd echoes will be used. The TR between two radio frequency pulses will be 200 ms, with no cardiac gating. In the liver, a large ROI will be chosen in a homogeneous area of the liver parenchyma without blood vessels. Analysis will then be performed as described for the heart multiecho.
Echocardiography indices
A detailed paediatric echocardiography will be performed to rule out any undiagnosed structural heart disease. This will be performed in the paediatric cardiology echocardiography unit, according to standard protocol using Philips IE33 or GE 9 machine with a 5–12 MHz phased array transducer.
The following measurements will be observed:
-
LV volume and systolic function: LV volumes and EF will be obtained from apical 4 chamber and short axis views using the 5/6×area×length method.37 These values will be compared to anthropometric based z-scores.
-
Diastolic function: Mitral and tricuspid E and A waves, mitral and tricuspid E/A ratios, mitral lateral and septal E′ tricuspid E′ and E/E′ ratios.
Speckle tracking
Longitudinal speckle tracking imaging derived strain peak (in %) will be obtained using the software offline package (TomTec Imaging Systems, Inc, Unterschleissheim, Germany) by tracing images obtained from the apical 4 chamber view (figure 3). Using the speckle-tracking algorithm, the package tracks a total of 49 points (ie, speckles) along the ventricular wall and interventricular septum (IVS). The ventricle is then divided into six segments, three along the free wall (basal, mid and apical free wall) and three along the IVS (basal, mid and apical septum), and the average of the speckles in the respective segments is displayed as the peak longitudinal strain for that segment. We will also calculate the mean peak longitudinal strain for the right ventricular (RV) and LV lateral free walls and IVS as an average of the three segmental values (base, mid and apex). The package also reports a global longitudinal strain peak for RV and LV. Since longitudinal fibres shorten during systole, longitudinal strain is reported as a negative value. Higher negative values reflect larger deformation.
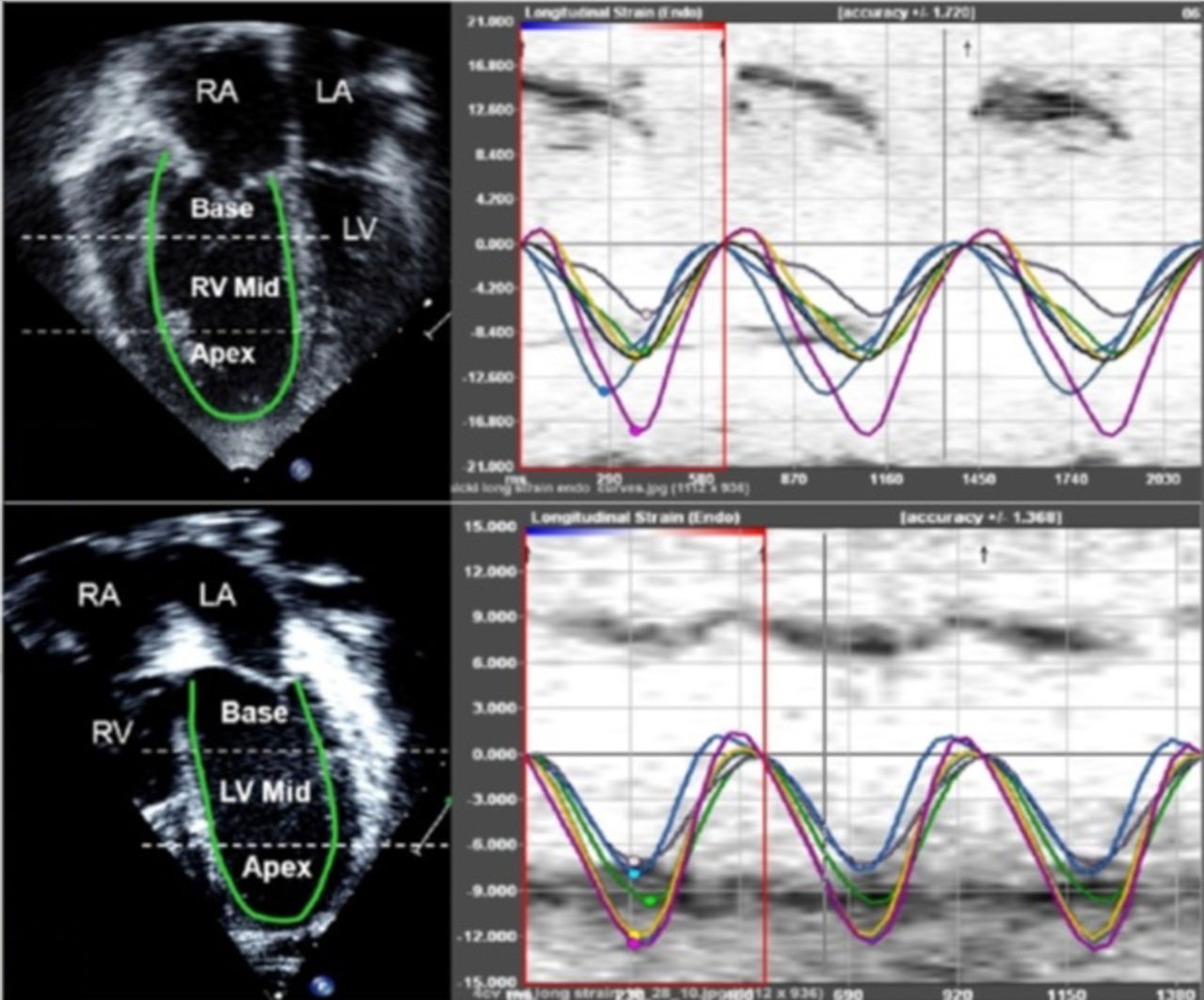
{kind=link}
{kind=link}
{kind=link}
Sample showing ventricular strain assessment using speckle tracking. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
Time to peak longitudinal strain (ms) will be calculated for three segments each on the LV free wall, IVS and RV free wall. Values for the three segments will be averaged to calculate the average time to peak strain for each wall (figure 3). The intraventricular RV delay will be calculated as the difference between RV free wall and IVS times to peak strain, LV intraventricular delay will be calculated as the difference between LV free wall and IVS times to peak strain, and the interventricular (between RV and LV) delay will be calculated as the difference between RV free wall and LV free wall times to peak strain.
Data collection: Data will be collected using abstraction forms. Adverse effects will also be collected using the AE form. These forms and data spreadsheets will be kept under lock and key. The computer library will be password protected. Any serious adverse effects associated with amlodipine and its use will be reported to the ERC and granting agency by the CTU and PI. amlodipine to the Ethics Review Committee (ERC) and granting agency will be the responsibility of the CTU and PI. These events will also be monitored by the Data Safety and Monitoring Board (DSMB).
Statistical analysis
Outcome variables
-
Our primary outcome is T2* values, which we will measure in milliseconds. A T2* recording will be taken at the initial visit, at 6 months and at 12 months.
-
Secondary outcomes will be looking at differences in absolute LV size, EF, strain and strain rate, diastolic function as mentioned earlier. We will also look at the liver iron content and rate of deposition in the liver.
Data will be entered in SPSS V.19. Continuous parameters will be presented as the mean with SD. However, since T2* is not normally distributed, we will report T2* as the geometric mean (antilog of the mean of the log data) and per cent coefficient of variation (CV—equivalent to the variance of the mean in log scale) following log transformation of data.
Mann Whitney U or Wilcoxon sign-rank test will be used for testing of continuous variables. Fisher exact or χ2 test will be used for categorical variables. A p value of <0.05 will be used as significance.
T2* calculations will be performed by two independent observers on 10 studies to account for interobserver variability. At least 10 out of all the 60 potential T2* cardiac MRIs and calculations (baseline and two follow-up visits included) will be sent to an international centre for validation. Similarly, myocardial strain analysis will also be performed by two observers on 10 studies to determine interobserver variability. One of the observers will recalculate the strain and strain rate on 10 studies after a week of the original measurements to help determine intraobserver variability.
Following completion of imaging at 6 months, an interim analysis will be performed.
To ensure proper conduct of the study along with the safety of participants and the validity and integrity of the data, all demographical and working details as well as data and statistical analysis will be reviewed by an independent DSMB. The DSMB will comprise of a statistician, a paediatric cardiologist and a haematologist and will be responsible for protecting the credibility of the trial by virtue of its independence from the staff involved in the trial, and will help to ensure the validity of study results by reviewing data on participant accrual and conducting independent interim reviews. A research steering committee comprising of a haematologist, a paediatrician and a paediatric health researcher will be created to ensure effective running of the study.
Publication and presentation
Timeline and study status
One year from approval. Patient recruitment will begin immediately after registration on clinicaltrials.gov and will continue for 4 months. The detailed study will be completed within 12 months of final recruitment and will be sent to a leading journal for publication.
We have currently completed the recruitment of patients for our project in September 2014. All recruited patients have undergone baseline imaging and pertinent laboratory testing and clinical examinations. The participants have been assigned to their respective arms with some of our earlier recruits near to completing the first 6 months of the study. As mentioned in the protocol, repeat imaging and serum ferritin levels will be obtained at 6 months. We anticipate our 6-month imaging to begin from December 2014 and to continue up to March 2015. On the basis of this timeline, we hope to complete our exit 12-month imaging from June 2015 to September 2015 and conclude our study by September 2015. As mentioned earlier, all our study patients will be reviewed in the clinics regularly and managed as per clinical needs.
Dissemination of results
Findings of this study will be reported through scientific publications and research conferences. Additionally, the findings will also be disseminated to the participants after project completion through a project summary and conclusion document distributed in an exit meeting and pertinent patient awareness sessions will be planned with the participants after the conclusion of the project. Additional information about our future work and study plans as well as clinical implications and future management regimens will also be discussed.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors BSH is the guarantor of the trial and responsible for the trial conception and design. BSH is also the contact person for all published versions of the protocol and manuscript. BSH, ZF, MZ and SC were involved in the conception of this trial. BSH, AS and FQ were involved in obtaining institutional approval. In addition to these authors, AS, ASh and NA were involved in the development of the protocol. AR was the study statistician. FAT performed patient MRIs. BSH performed patient echocardiography. ASh and NA were responsible for patient recruitment. NA was involved in data collection and maintenance of trial data and patient supervision. AR, NA and AS conducted data analysis. In addition to these authors, BSH also assisted in data interpretation. The final manuscript was drafted with the input and approval of all authors. Throughout the trial, BSH, ZF, SC, FAT, MK and ZS provided expert advice as well as policy and consumer perspectives.
-
Funding Grand Challenges Canada (grant number: S40270-01).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This study has been approved by the Ethical Review Committee at the Aga Khan University Hospital that will also serve as an independent data monitoring committee for this pilot research trial.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All unpublished data are exclusively in the custody of the principal investigator.