Article Text

Abstract
Introduction The management of recurrent malignant pleural effusions (MPE) can be challenging. Various options are available, with the most efficacious and widely used being talc pleurodesis. Talc can either be applied via a chest drain in the form of slurry, or at medical thoracoscopy using poudrage. Current evidence regarding which method is most effective is conflicting and often methodologically flawed. The TAPPS trial is a suitably powered, multicentre, open-label, randomised controlled trial designed to compare the pleurodesis success rate of medical thoracoscopy and talc poudrage with chest drain insertion and talc slurry.
Methods and analysis 330 patients with a confirmed MPE requiring intervention will be recruited from UK hospitals. Patients will be randomised (1:1) to undergo either small bore (<14 Fr) Seldinger chest drain insertion followed by instillation of sterile talc (4 g), or to undergo medical thoracoscopy and simultaneous poudrage (4 g). The allocated procedure will be performed as an inpatient within 3 days of randomisation taking place. Following discharge, patients will be followed up at regular intervals for 6 months. The primary outcome measure is pleurodesis failure rates at 3 months. Pleurodesis failure is defined as the need for further pleural intervention for fluid management on the side of the trial intervention.
Ethics and dissemination The trial has received ethical approval from the National Research Ethics Service Committee North West—Preston (12/NW/0467). There is a trial steering committee which includes independent members and a patient and public representative. The trial results will be published in a peer-reviewed journal and presented at international conferences, as well as being disseminated via local and national charities and patient groups. All participants who wish to know the study results will also be contacted directly on their publication.
Trial registration number ISRCTN47845793.
- Pleurodesis
- Malignant pleural effusion
- Randomised controlled trial
- Talc
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
Suitably powered multicentre, randomised controlled trial of talc pleurodesis interventions in the general malignant pleural effusion population.
-
First study to specifically investigate poudrage using medical thoracoscopy.
-
Robust 6-month patient follow-up.
-
Clinically relevant and applicable definition of pleurodesis success.
-
Pleurodesis performed as part of diagnostic thoracoscopy not included.
-
No comparison with indwelling pleural catheters.
Introduction
Pleural effusions are a common complication of many cancers, with symptoms often requiring intervention. Data from 10 years ago suggest that there are up to 175 000 new cases of malignant pleural effusion (MPE) in the USA per year and around 40 000 cases per year in the UK,1 although these figures may now be conservative as the global burden of malignancy continues to rise each year, and with it the incidence of MPE.
Pleurodesis is the adherence of the visceral and parietal pleura, which causes an obliteration of the pleural space. Removing the pleural space reduces the possibility of pleural fluid build-up, which means that induction of pleurodesis is considered the mainstay of treatment for recurrent MPE. Many substances have been shown to induce chemical pleurodesis, although by far the most commonly used one in Europe and North America is talc, which has been shown to be superior to alternatives such as tetracycline or bleomycin.2 Overall, pleurodesis success rates with talc are typically high, ranging from 81% to 100%,3 although this efficacy may vary considerably in real-world practice due to differences between clinicians and individual centres. The traditional method to instil talc, the control arm in this study, requires a patient to be admitted to hospital for chest tube insertion and fluid drainage. Talc is administered as slurry and is made up with a physiologically inert fluid such as 0.9% saline. The chest tube is removed once subsequent drainage volumes become low, potentially indicating successful pleurodesis.
An alternative to this approach is the application of sterile talc powder under direct vision at thoracoscopy (insufflation or poudrage). However, despite an increasing number of hospitals having access to medical thoracoscopy, it is still much less ubiquitous than Seldinger chest drain insertion, with the requirement for specialist training and the increased costs of the procedure being major limitations, along with the more complex nature of the procedure. The efficacy of talc poudrage at 1 month for pleurodesis has been documented in a number of studies. Published success rates tend to lie around 85%, although there is significant heterogeneity between study groups limiting reliability. A number of studies regarding talc slurry and talc poudrage were compared as part of the 2004 Cochrane review which, along with suggesting talc was the most efficacious sclerosant, found talc poudrage at thoracoscopy to have an improved relative risk of non-recurrence (1.19) over talc slurry.2 A subsequent large randomised trial by Dresler, published in 2005, suggested there was only a trend towards superiority of poudrage (p=0.1), with no significant overall difference between the two methods. Post hoc subgroup analysis demonstrated a rise in pleurodesis success once patients with trapped lung were excluded, as well as a significant difference between poudrage (82%) and slurry (71%; p=0.045).4
The role of talc poudrage for the induction of pleurodesis and the prevention of fluid recurrence in MPE remains unclear. Chest drain insertion with talc slurry is universally available, less expensive and relatively easy to perform, but may have a significantly poorer success rate and may result in longer hospital stays. The TAPPS trial aims to definitively resolve the question of whether talc poudrage is a superior method for the induction of pleurodesis in MPE, allowing clinicians to make the most appropriate and best informed decisions and recommendations to patients.
Methods and analysis
This study, evaluating the efficacy of thoracoscopy and talc poudrage versus pleurodesis using talc slurry (TAPPS trial), is a multicentre, open-label, randomised controlled trial. The trial is sponsored by the North Bristol NHS Trust (NBT) and coordinated jointly by the Academic Respiratory Unit at the University of Bristol and the Oxford Respiratory Trials Unit (ORTU) at the University of Oxford. Data management is undertaken by the ORTU. The trial is registered on the International Standardised Randomised Controlled Trial Registry (ISRCTN47845793) and funded by the National Institute for Health Research (NIHR) Health Technology Assessment programme. The study is included in the NIHR Clinical Research Network portfolio (ID: 12537). The trial will be conducted in accordance with the Declaration of Helsinki and good clinical practice (GCP).
The primary research question is: For patients with a confirmed MPE and good performance status, does thoracoscopy and talc poudrage increase the proportion of patients with successful pleurodesis at 3 months postprocedure, when compared with standard therapy with chest drain insertion and talc slurry instillation?
The secondary research questions are:
-
Does talc poudrage reduce the time to pleurodesis failure at 3-month and 6-month postprocedure when compared with talc slurry?
-
Does talc poudrage at thoracoscopy improve chest X-ray (CXR) appearances after initial drain removal, and at 1-month, 3-month and 6-month postrandomisation when compared with talc slurry?
-
Does talc poudrage cause less breathlessness and thoracic pain for the first 7 days postrandomisation when compared with talc slurry?
-
Does talc poudrage improve health-related quality of life over the 6 months of postrandomisation when compared with talc slurry?
-
Is talc poudrage cost-effective over 6 months when compared with talc slurry instillation?
-
Does talc poudrage reduce healthcare utilisation during the 6-month postrandomisation when compared with talc slurry instillation?
Setting
Three hundred and thirty patients requiring a pleurodesis intervention for a confirmed MPE will be recruited from UK hospitals (see online supplementary appendix 1 for details of recruiting centres). Patients will be randomised to undergo either chest drain insertion followed by 4 g talc slurry instillation, or to undergo medical thoracoscopy with 4 g talc poudrage. The study flow diagram is shown in figure 1.

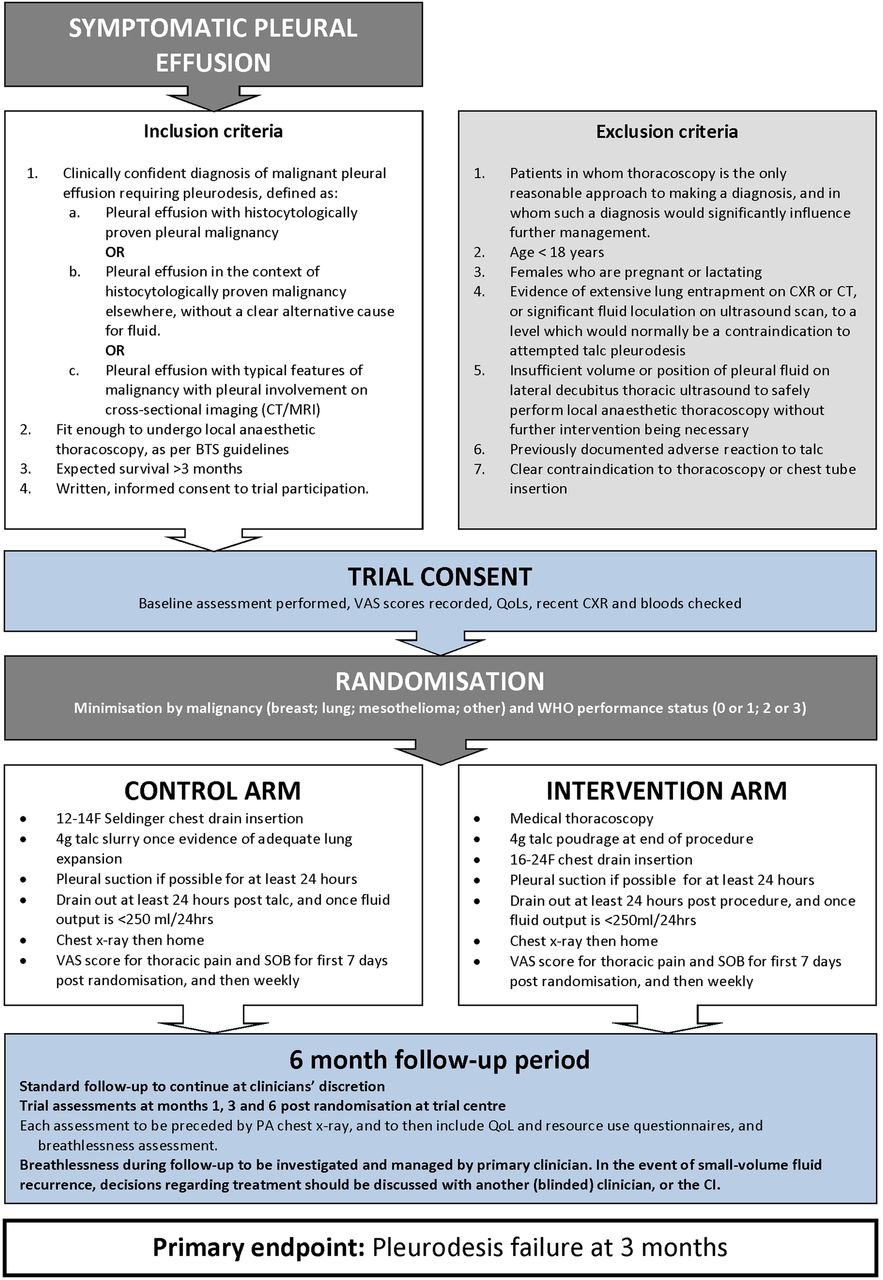{kind=link}
Trial flow chart (BTS, British Thoracic Society; CI, chief investigator; CXR, chest X-ray; QoL, quality of life; VAS, visual assessment scale; PA, pleural apposition; SOB, shortness of breath).
Subject screening and selection
Patients with MPE will be identified following early discussion at each centre's cancer multidisciplinary team meetings (MDT), at routine outpatient appointments and during inpatient reviews. Eligible patients will be invited to participate on a consecutive basis, and will be provided with a patient information leaflet at the earliest opportunity (see online supplementary appendix 2). Patients can be enrolled only once into the TAPPS trial.
Inclusion criteria
-
Clinically confident diagnosis of MPE requiring pleurodesis, defined as:
-
Pleural effusion with histocytologically proven pleural malignancy; or
-
Pleural effusion in the context of histocytologically proven malignancy elsewhere, without a clear alternative cause for fluid; or
-
Pleural effusion with typical features of malignancy with pleural involvement on cross-sectional imaging without a clear alternative cause for fluid.
-
-
Fit enough to undergo local anaesthetic thoracoscopy.
-
Expected survival >3 months.
-
Written informed consent to trial participation.
Exclusion criteria
-
Patients in whom thoracoscopy is the only reasonable approach to making a diagnosis, and in whom such a diagnosis would significantly influence further management;
-
Age <18 years;
-
Females who are pregnant or lactating;
-
Evidence of extensive lung entrapment on CXR or a CT scan, or significant fluid loculation on an ultrasound scan, to a level which would normally be a contraindication to attempted talc pleurodesis;
-
Insufficient volume or position of pleural fluid on lateral decubitus thoracic ultrasound to safely perform local anaesthetic thoracoscopy without further intervention being necessary;
-
Previously documented adverse reaction to talc;
-
Clear contraindication to thoracoscopy or chest tube insertion.
Informed consent
A doctor will confirm patient eligibility prior to consent being taken. Participation in the trial will be discussed with the patient by a medical or nursing member of the local trial team. Patients will be given sufficient time (in their own opinion) to fully consider trial entry, as well as to ask questions of investigators. The consent form (see online supplementary appendix 3) will be countersigned by either a medical or nursing member of the trial team.
Randomisation
Following informed consent, patients will be randomly assigned in a 1:1 ratio using minimisation with a random element to undergo either chest drain insertion with talc slurry pleurodesis or thoracoscopy with talc poudrage. The day of randomisation is defined as day 0. Although the allocated trial procedure may be performed within 3 days of randomisation, every effort should be made to perform the procedure immediately afterwards.
Treatment allocation will be performed over the telephone by the ORTU. The randomisation sequence will be generated using a validated, online randomisation service (Sealed Envelope, London, UK; http://www.sealedenvelope.com).
The minimisation factors are:
-
Type of underlying malignant disease (mesothelioma, lung cancer, breast cancer, other);
-
WHO/Eastern Cooperative Oncology Group (ECOG) performance status (0 or 1; 2 or 3).
Patients and clinicians will not be blinded to treatment allocation.
Standard care
All patients should have been discussed in their local or regional tumour-specific MDT. For all issues other than those pertaining to the drainage and management of the MPE, treatment discretion lies with the primary clinician.
Normal clinical review during the trial period will take place in the usual outpatient or inpatient setting, and will typically be carried out by oncologists or respiratory physicians. The frequency of clinical review will depend on patient choice, severity of symptoms and clinical discretion. In general, patients who are managed with chemotherapy for underlying malignancy are typically reviewed every 2–3 months.
Patients can withdraw from the trial at any time without their clinical care being affected.
Co-enrolment in other clinical trials will be discussed on an individual patient basis, but patients should not be co-enrolled into any trial which specifically aims to directly influence pleural fluid production or drainage.
Interventions
The full trial specific procedures (TSP) for the two treatment arms can be found in online supplementary appendices 4 and 6.
Control (talc slurry) arm
Patients will have a small-bore (<14 Fr) chest drain inserted under aseptic conditions using the Seldinger technique, with appropriate local anaesthesia and premedication as necessary. A suitable site for drain placement will be identified using contemporaneous ultrasound. Drains will only be inserted by persons with adequate training and experience. Trial pleural fluid samples (see section below) should also be taken as necessary.
A CXR should be performed between 18 and 24 h after drain insertion. If there is no evidence of trapped lung or significant fluid, as determined by the patient's primary physician, then the patient should have 4 g talc slurry instilled through the chest drain, following the appropriate TSP. Patients who continue to have evidence of significant pleural opacification may need to undergo further imaging to confirm the cause. If the significant component of the opacification is felt to be due to pleural thickening rather than fluid, then slurry instillation should proceed according to the TSP.
Patients who have evidence of trapped lung, or who have significant opacification due to fluid on CXR, may have thoracic suction applied if it is felt appropriate. Patients should undergo slurry instillation once the primary physician is satisfied that at least 50% of the visible pleura are apposed. If, by 48 h post drain insertion, there is inadequate pleural apposition on CXR, or the primary physician feels that talc slurry instillation would be inappropriate for another reason, then further management decisions lie with the primary physician. Such patients should continue to receive follow-up in the standard manner and should have all treatment decisions clearly documented. A flow chart for patient management in the control arm is provided (see online supplementary appendix 5).
Following slurry instillation, thoracic suction should be applied if available and tolerated. Once documented drainage falls below 250 mL per 24 h (in the presence of a patent drain), the drain should be removed, unless the primary physician feels there is reason for the drain to remain in place for longer. Following drain removal, a further CXR should be performed and an appointment given for the first trial follow-up visit at 1-month postrandomisation.
Intervention (talc poudrage) arm
All participants who undergo thoracoscopy will have their procedure performed by persons with adequate training and experience. Patients will be given adequate sedation (if required) and local anaesthetic for the procedure. Biopsy samples will be taken as needed. Trial pleural fluid samples (see section below) should also be taken as necessary. At the end of the procedure, 4 g of sterile talc should be sprayed over the pleural surfaces. A 16–24 Fr chest drain should be inserted at the end of the procedure and connected to an underwater seal. Patients should be attached to thoracic suction, if available and tolerated.
The future care decisions of any patient whose procedure is abandoned or curtailed before poudrage is performed (at the discretion of the operator) remain with the primary physician. Such patients will remain under trial follow-up and should have all care decisions and associated delays clearly documented in their notes.
A CXR should be performed between 18 and 24 h after drain insertion to assess lung re-expansion. If there is evidence of incomplete re-expansion, then drain patency should be checked. The management of patients with incomplete lung expansion is at the discretion of the primary physician, and may include the continued use of thoracic suction.
All patients’ drains should remain in place for a minimum of 24 h. When a patient has drained 250 mL or less in the previous 24 h, then the drain should be removed, unless the primary physician feels that it needs to remain in place for longer. A flow chart for patient management in the intervention arm is provided (see online supplementary appendix 7). Following drain removal, a further CXR should be performed and an appointment given for the first trial follow-up visit at 1-month postrandomisation.
Data collection and management
Visual assessment scale (VAS) scoring
All patients will document a VAS score for thoracic pain and breathlessness during their baseline assessment. This score should then be performed again on the first day postrandomisation, and then daily for 7 days. Following this, scores should be completed on a weekly basis.
Patient diaries
Patients will be provided with preprinted diaries. They are to record all personal contact with medical professionals (excluding trial visits) in a basic standardised manner. These data will be reviewed at follow-up appointments and will subsequently be used to determine the health utilisation of each participant during the follow-up period.
Biological samples and storage
At all trial sites, those who consent to trial sample analysis should have 2 EDTA tubes, 1 serum gel tube and 1 lithium heparin tube of blood taken (‘trial blood samples’). Sites other than Oxford and North Bristol should send these samples as soon as possible, unprocessed, to the Respiratory Research Unit at Southmead Hospital. Patients at North Bristol and Oxford should also have 2 EDTA, 1 serum gel and 1 lithium heparin tube filled with pleural fluid during either thoracoscopy or initial drain insertion (‘trial pleural fluid samples’). At these sites, trial blood and pleural fluid samples should be centrifuged, labelled and stored locally initially as per the appropriate TSP. All processed samples will eventually be transferred to the Respiratory Research Unit at North Bristol. Genetic compositional analysis may also be undertaken on participants’ samples if specific consent for this has been obtained.
Additionally, on the second day post talc administration (or on discharge if sooner), patients should have blood samples taken and analysed locally for C reactive protein, full blood count, and urea and electrolytes, with the results entered onto the discharge case report form.
Trial follow-up appointments
Trial follow-up appointments will take place at 1-month, 3-month and 6 month postrandomisation, with telephone follow-ups being performed if necessary. A CXR will be performed and patients will undergo a standardised assessment, including a review of their healthcare resource use diary; EQ-5D and SF-36 quality of life questionnaires; and a focused medical history.
Further pleural intervention
All patients who are felt to have increasing breathlessness should undergo a CXR. Any CXR which shows a degree of pleural opacification ipsilateral to the pleurodesis attempt should lead to further imaging to confirm the presence of fluid. If fluid is confirmed, and the CXR shows pleural opacification to be one-third or greater than the volume of the hemithorax (by visual estimation), the primary physician should undertake any further investigations or interventions as deemed appropriate. In patients who have less than one-third of the hemithorax occupied by pleural fluid, the primary physician should discuss with another local physician who is blinded to the treatment arm whether pleural intervention is required.
Data management
Clinical Record Forms (CRF) will be completed by the trial team at recruiting centres and sent to the ORTU. Data will then be entered onto the trial database (OpenClinica clinical trials software). Missing data and data queries will be highlighted to the trial teams on a monthly basis. The CRFs will only identify patients using their personal trial identification number (no identifiable patient information).
Primary outcome
The primary outcome is the number of patients who experience pleurodesis failure up to 3 months (90 days) postrandomisation.
A patient is defined as experiencing pleurodesis failure if they undergo any of the following procedures on the side ipsilateral to their trial intervention:
-
Therapeutic pleural aspiration of ≥100 mL; or
-
Insertion of an intercostal drain for fluid drainage; or
-
Insertion of an indwelling pleural catheter; or
-
Medical or surgical thoracoscopy.
A patient is also deemed to have failed pleurodesis if their primary physician decides that they require one of the above pleural interventions, but the intervention is not performed. The primary physician is not blind to the treatment arm; however, all decisions to intervene or not in effusions which occupy less than or equal to one-third of the hemithorax will be discussed with a second clinician who is blind to treatment allocation.
Secondary outcomes
The trial's secondary outcomes are:
-
The number of patients with pleurodesis failure up to 30 days postrandomisation.
-
The number of patients with pleurodesis failure up to 180 days postrandomisation.
-
Requirement for further pleural procedures up to 180 days postrandomisation, based on an independent assessment performed by two adjudicators who are blind to the treatment outcome and clinical course.
-
Percentage pleural opacification (on CXR) at 1-month, 3-month and 6-month postrandomisation follow-up visits, and after initial drain removal.
-
Self-reported health-related quality of life at 1-month, 3-month and 6-month follow-up postrandomisation visits, measured using SF-36 and EQ-5D questionnaires.
-
Self-reported thoracic pain and breathlessness (postrandomisation) at 7, 30, 90 and 180 days, measured using VAS scores.
-
All-cause mortality up to 180 days postrandomisation.
-
Time to pleurodesis failure, censored at 180 days postrandomisation.
-
Number of nights spent in the hospital up to 90 days postrandomisation, including length of initial hospital stay.
Sample size calculation
Previous literature and our own audit data suggest that patients with a WHO performance status score of 2 or better have approximate pleurodesis failure rates of ≤10% with thoracoscopy, and ≥30% with a ‘best standard of care’ standard chest tube and talc slurry pleurodesis.5
In order to detect a ≥15% difference in pleurodesis failure at 3 months (10% thoracoscopy and poudrage vs 25% chest drain and talc slurry) with 90% power, a 5% significance level and 10% loss to follow-up, the study requires 325 patients. For the present analysis, numbers have been rounded up to include 330 patients (165 patients in each treatment arm).
Statistical analysis plan
The full statistical analysis plan is published elsewhere.
The primary analysis for each outcome will be by intention to treat. All tests will be two-sided, and will be considered statistically significant at the 5% level. For each analysis, the following summaries will be provided:
-
The number of patients in each treatment group who are included in the analysis.
-
The mean (SD) or median (IQR) in each treatment group for continuous outcomes, or the number (%) of patients experiencing an event for binary or time-to-event outcomes (time-to-event outcomes will also present the median time to event in each treatment arm if applicable).
-
The treatment effect (difference in means for continuous outcomes, OR for binary outcomes, HR for time-to-event outcomes, rate ratio for count outcomes) with its 95% CI and a p value.
All analyses will adjust for minimisation variables (type of underlying malignant disease (mesothelioma, lung cancer, breast cancer, other) and WHO performance status (0–1 or 2–3)).6–9 The minimisation variables will be included as covariates in the regression model for each outcome.
CONSORT data will be presented, including: the number of patients screened for the study; the numbers randomised; the numbers receiving the interventions; the numbers lost to follow-up and excluded (with reasons) and the number of patients included in the primary analysis.
Subgroup analyses will be performed for the primary outcome, and the following secondary outcomes: pleurodesis failure at 30 and 180 days; requirement for further pleural procedures; and percentage CXR opacification. Results from subgroup analyses will be viewed as hypothesis generating, and will not be used to make definitive statements about treatment efficacy in a specific subgroup of patients. The following subgroup analyses will be performed:
-
Patients receiving anticancer therapy at baseline versus those not receiving;
-
Previous radiotherapy to chest versus no previous radiotherapy to chest;
-
WHO performance status 0–1 versus 2–3;
-
Patients on non-steroidal anti-inflammatory drugs (NSAIDS) at baseline versus those not on NSAIDS at baseline;
-
Patients on steroids at baseline versus those not on steroids at baseline;
-
Previous attempt at pleurodesis within the past month versus no attempt in the past month;
-
Patients with primary malignancy of breast cancer versus mesothelioma versus lung cancer versus other.
Changes to the protocol after trial commencement
The trial details documented here are consistent with the TAPPS Trial protocol V.6 (date: 06/10/2014). A summary of the trial amendments can be found in online supplementary appendix 8.
In September 2013, the ‘window’ in which individuals could undergo their allocated trial procedure was extended from 24 to 72 h postrandomisation.
End of trial
The trial will end once 330 patients have been recruited and all patients have died or completed 6 months of trial follow-up (whichever is sooner).
Ethics and dissemination
Monitoring
An independent data monitoring committee (IDMC) will be convened at regular intervals, consisting of members who are independent of the trial investigators. The role of the IDMC is to review study safety data and provide advice to the trial steering committee (TSC), specifically as to whether recruitment can continue. No interim analysis is planned.
Safety reporting
Data will be collected at each patient's trial visit regarding any serious adverse events (SAE; as defined by GCP). All SAEs causally related to trial interventions will be reported to the sponsor and to the relevant oversight bodies, and will be followed until they resolve or stabilise.
Trial monitoring and oversight
The TSC will be responsible for overseeing the progress of the trial and will meet at approximate six monthly intervals. The TSC will comprise of independent chairperson, independent members, statistician, patient and public representative and members of the trial team.
Dissemination
The trial will be publicised at regional and national conferences. The final results will be presented at scientific meetings and published in a peer-reviewed journal (authorship will be according to the journal's guidelines). In addition, a lay summary of the study results will be circulated to potentially interested parties.
Acknowledgments
The authors are grateful for the infrastructure provided by the local Cancer Research Networks, and to all the trial teams involved in patient recruitment (see online supplementary appendix 1).
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online appendix 1
- Data supplement 2 - Online appendix 2
- Data supplement 3 - Online appendix 3
- Data supplement 4 - Online appendix 4
- Data supplement 5 - Online appendix 5
- Data supplement 6 - Online appendix 6
- Data supplement 7 - Online appendix 7
- Data supplement 8 - Online appendix 8
Footnotes
-
Contributors NAM and NMR conceived the initial trial concept. All authors contributed to the development of the trial design and protocol. NMR and BCK carried out the sample size calculations. RB, BCK, NMR, RFM and NAM wrote the statistical analysis plan. All authors have read and approved this manuscript.
-
Funding This trial is supported by The National Institute for Health Research (NIHR) Health Technology Assessment (HTA) programme; project number 10/50/42.
-
Competing interests None.
-
Ethics approval The trial and all substantial amendments have been reviewed and granted approval by the National Research Ethics Service (NRES) Committee North West—Preston (12/NW/0467).
-
Provenance and peer review Not commissioned; peer reviewed for ethical and funding approval prior to submission.