Article Text

Abstract
Objective To evaluate the clinical utility of individualising dual antiplatelet therapy (DAPT) after percutaneous coronary intervention (PCI) in an all-comers population, including ST-elevation myocardial infarction (STEMI) patients.
Setting Tertiary care single centre registry.
Participants 1008 consecutive PCI patients with stent implantation, without exclusion criteria.
Intervention Peri-interventional individualisation of DAPT, guided by multiple electrode aggregometry (MEA), to overcome high on-treatment platelet reactivity (HPR) to ADP-induced (≥50 U) and arachidonic acid (AA)-induced aggregation (>35 U).
Outcome measures The primary efficacy end point was definite stent thrombosis (ST) at 30 days. The primary safety end point was thrombolysis in myocardial infarction (TIMI) major and minor bleeding. Secondary end points were probable ST, myocardial infarction, cardiovascular death and the combined end point: major cardiac adverse event (MACE).
Results 53% of patients presented with acute coronary syndrome (9% STEMI, 44% non-ST-elevation). HPR to ADP after 600 mg clopidogrel loading occurred in 30% of patients (73±19 U vs 28±11 U; p<0.001) and was treated by prasugrel or ticagrelor (73%), or clopidogrel (27%) reloading (22±12 U; p<0.001). HPR to ADP after prasugrel loading occurred in 2% of patients (82±26 U vs 19±10 U; p<0.001) and was treated with ticagrelor (34±15 U; p=0.02). HPR to AA occurred in 9% of patients with a significant higher proportion in patients with HPR to ADP (22% vs 4%, p<0.001) and was treated with aspirin reloading. Definite ST occurred in 0.09% of patients (n=1); probable ST, myocardial infarction, cardiovascular death and MACE occurred in 0.19% (n=2), 0.09% (n=1) and 1.8% (n=18) of patients. TIMI major and minor bleeding did not differ between patients without HPR and individualised patients (2.6% for both).
Conclusions Individualisation of DAPT with MEA minimises early thrombotic events in an all-comers PCI population to an unreported degree without increasing bleeding. A randomised multicentre trial utilising MEA seems warranted.
Trial registration number http://www.clinicaltrials.gov; NCT01515345.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
The strengths of our study are: first, the real world percutaneous coronary intervention setting with inclusion of every consecutive patient with stent implantation, without any exclusion criteria; second, the consequent and efficient peri-interventional individualisation of dual antiplatelet therapy, with only 0.3% of patients left on high on-treatment platelet reactivity to ADP at the time of hospital discharge; and third, the minimisation of ischaemic events within 30 days by nearly abolishing early definite stent thrombosis, without increasing bleeding complications.
-
Limitations of our study are the non-randomised and monocentric registry design without control group concerning ischaemic events.
Introduction
High on-treatment platelet reactivity (HPR) to ADP represents one of the strongest independent risk factors for postpercutaneous coronary intervention (PCI) ischaemic events in patients given dual antiplatelet therapy (DAPT), according to numerous observational studies using various platelet function tests.1–3
Whether HPR represents only a marker of higher risk or a modifiable risk factor is still a matter of debate,2 as prospective randomised trials evaluating personalised antiplatelet therapy aiming to overcome HPR resulted in conflicting data. Smaller randomised trials,4 as well as non-randomised studies5 and a recent meta-analysis6 suggested a significant clinical benefit, but three randomised studies failed to do so.7–9 However, each of these trials, utilising the VerifyNow assay, was afflicted with major limitations potentially masking the real value of individualising DAPT after PCI in daily practice.1 ,10 Their low-risk population and primarily the high selection bias in GRAVITAS7 and TRIGGER-PCI,9 with patient inclusion more than 12 h after PCI, seem to cloud the potential importance of optimising platelet inhibition at the time of PCI. By contrast, the very recent CHAMPION Phoenix trial11 provides a more realistic scenario of expectable ischaemic complications during and after PCI. More than 11 000 patients with oral clopidogrel loading, including the whole clinical PCI spectrum (56% stable coronary artery disease (CAD), 26% non-ST-elevation acute coronary syndrome (NSTE-ACS), 18% ST-elevation myocardial infarction (STEMI)), were preinterventionally randomised to receive an intravenous bolus and infusion of cangrelor, a fast acting reversible ADP receptor blocker. Ischaemic complications in the whole study cohort occurred in 5.3%, including a definite stent thrombosis (ST) rate of 1.1% during the first 48 h. Notably, the majority of events occurred within 6 h after PCI.
HPR to acetylic salicylic acid (ASA) is less well studied and its clinical relevance is unclear. The ADAPT-DES registry3 found no difference in response to ASA, measured by the VerifyNow assay, between patients with and without ST. Data not only from our group, however, suggested that dual HPR to ADP-induced as well as arachidonic acid-induced (AA; reflecting response to ASA) aggregation, measured by multiple electrode aggregometry (MEA)12 or the VerifyNow assay,13 predisposes patients to a higher ischaemic risk than single HPR. Furthermore, MEA has been shown to effectively assess the risk of HPR to ADP after PCI14 with higher accuracy than the vasodilator-stimulated phosphoprotein phosphorylation assay15 utilised in the Bonello studies.
Therefore, our registry aimed to evaluate the impact of individualising DAPT with MEA in an all-comers population, including STEMI patients without exclusion criteria, by peri-interventional treatment of HPR to ADP and AA.
Methods
Patient population
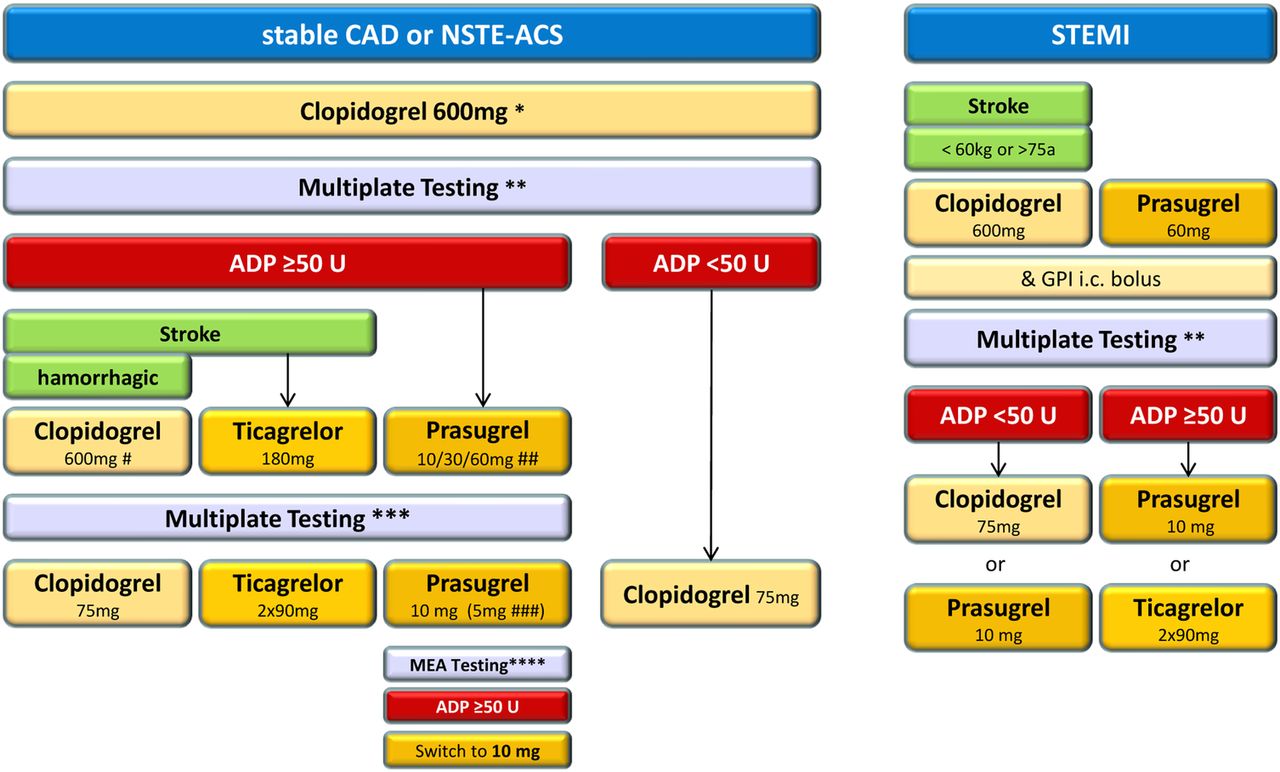
This was a prospective, single-centre cohort observation of consecutive PCI patients, including all forms of ACS (including cardiogenic shock) and all stable CAD, with stent implantation or drug eluting balloon dilatation (for treatment of instent restenosis), and without exclusion criteria (secondary causes for ACS, like anaemia had to be corrected according to standard patient care, but did not represent an exclusion criterion, nor did thrombocytopenia or liver dysfunction once the indication for an invasive approach was given). Patients without stent implantation (ie, unsuccessful reopening of a chronic total occlusion or balloon dilatation only) were not included. Peri-interventional individualisation of platelet inhibition was performed according to the protocol shown in figure 1 and described in detail below. Informed consent was obtained after PCI, either from the patient or from the guardian in cases of critically ill conditions. Follow-up information was obtained by either direct outpatient visit or telephone contact at 30 days.

Algorithm of ADP receptor blocker treatment. CAD, coronary artery disease; GPI, glycoprotein IIbIIIa inhibitor; MEA, multiple electrode aggregometry; NSTE-ACS, non-ST-elevation acute coronary syndrome; STEMI, ST-elevation myocardial infarction. *Loading in stable patients the day before angiography; **platelet testing not earlier than 12 h after loading, and at the latest at the time of diagnostic angiography, after GPI administration serial testing up to 7 days; ***platelet testing the day after reloading; ****platelet testing 1 week after starting 5 mg prasugrel; #up to three clopidogrel reloadings; ##prasugrel reloading dependent on residual reactivity: ADP >80: 60 mg, ADP 60–79: 30 mg, ADP 50–59: 10 mg; ###in patients <60 kg and/or >75 years.
Study end points
The primary efficacy end point was definite ST during 30 days follow-up. The secondary efficacy outcome parameters were probable ST, myocardial infarction and cardiovascular death, as well as a combination of the aforementioned end points as major cardiac adverse events (MACEs). Definite and probable STs were defined according to the Academic Research Consortium (ARC)16 and diagnosed by the authors without blinded adjudication. The primary safety end point was the incidence of thrombolysis in myocardial infarction (TIMI) bleeding complications.17 TIMI major bleeding was defined as intracranial bleeding or overt bleeding with a decrease in haemoglobin ≥5 g/dL. TIMI minor bleeding was defined as observed bleeding with decrease in haemoglobin ≥3–<5 g/dL.
Individualisation of DAPT
Individualisation of ADP receptor blocker treatment was performed according to the algorithm presented in figure 1. After an initial clopidogrel loading dose of 600 mg, on-treatment platelet reactivity was measured the next day by MEA, at the earliest after 12 h and at the latest at the time of diagnostic angiography. HPR was defined as ≥50 U ADP-induced aggregation. This cut-off represents the mean of published data from Sibbing and our group.14 ,15 From November 2008 to May 2009, patients with HPR were reloaded with clopidogrel 600 mg up to three times according to the Bonello protocol.4 After prasugrel18 became available in June 2009, HPR to clopidogrel was treated with prasugrel (Efient/Effient) loading, depending on the degree of the residual ADP-induced platelet reactivity: cases with ADP >80 U received 60 mg, ADP 60–79 U 30 mg and ADP 50–59 U 10 mg of prasugrel. This staged approach was chosen in order to avoid potential bleeding complications due to the observed over-response (ie, very ‘flat’ ADP and ASPI curves, <10–15 U) after a routine prasugrel 60 mg loading in patients with borderline clopidogrel response (ADP 50–60 U). In patients older than 75 years or weighing less than 60 kg, the maintenance dose (MD) of prasugrel was reduced to 5 mg according to the manufacturer's specification, with MEA testing 1 week later and dose adjustments, if necessary. In cases of contraindications to prasugrel (history of stroke), clopidogrel reloadings were performed, until ticagrelor (Brilique/Brilinta) became available. STEMI patients younger than 75 years and weighing more than 60 kg without history of stroke were primarily loaded with 60 mg prasugrel due to the local standard operating procedure of the Viennese STEMI network. After ticagrelor19 became available in March 2011, HPR to prasugrel and HPR to clopidogrel in patients with contraindications to prasugrel were treated with 180 mg ticagrelor loading. In cases of contraindications to ticagrelor (history of intracranial haemorrhage), clopidogrel reloadings were performed. Special care was taken to limit the possibility of HPR at the time of PCI by clopidogrel loading at least 12 h prior to PCI, with reloading, if necessary, either prior to PCI in case MEA testing was already known, or at the latest 1–2 h after PCI. In case of no oral ADP receptor blocker loading, or only within 4-6 h, pre-PCI was given (eg, STEMI or urgent invasive non-STEMI (NSTEMI) patients), bolus-only administration of a glycoprotein IIbIIIa inhibitor (GPI) (intracoronary abciximab (0.25 mg/kg; Reopro) or intravenous eptifibatide (180 µg/kg, Integrilin)) was performed. Thereafter, serial MEA measurements were taken for up to 7 days to allow determination of the level of oral ADP receptor inhibition. Details of this blocking and bridging strategy have been published previously.20 At discharge, all patients should be within the therapeutic range of platelet inhibition (ie, non-HPR).
Individualisation of ASA treatment was conducted as follows. Stable patients without chronic ASA treatment were loaded with 300 mg ASA orally once the day before angiography. Patients with ACS were loaded with ASA intravenous: 500 mg was used in ASA naïve patients and 250 mg was used in cases of chronic ASA treatment. HPR to ASA was defined as >35 U AA-induced aggregation. This cut-off represents a mean derived from published data12 ,21 and the MEA manufacturer's recommendations. ASA reloading was performed with either 300 mg orally once or 250 mg intravenous. In cases of HPR to ADP and ASA, first ADP receptor blocker reloading was performed with ASA reloading if necessary after MEA testing the next day.
PCI was performed according to the current standard guidelines. The type of stent implanted was at the discretion of the interventional cardiologist. In cases of drug eluting stent (DES) implantation, only second generation DESs were used (Biolimus-eluting: Biomatrix; Everolimus-eluting: Promus Element and Xience; Zotarolimus-eluting: Resolute). All patients received 100 IU/kg of unfractionated heparin, with adjustments according to measurements of activated clotting time, except in cases of GPI bolus administration, where only 70 IU/kg were given.
Impedance aggregometry
Whole blood aggregation was determined using MEA, a new-generation impedance aggregometer (Multiplate Analyzer, Roche, Munich, Germany). The system detects electrical impedance change due to the adhesion and aggregation of platelets on two independent electrode-set surfaces in the test cuvette, with a low rate of intra-assay and interassay variability.22 ADP and AA were used as agonists. A 1:2 dilution of whole blood anticoagulated with hirudin and 0.9% NaCl was stirred at 37°C for 3 min in the test cuvette. ADP (6.4 µM) and AA (0.5 mM) were added, and the increase in electrical impedance was continuously recorded for 6 min. The mean values of the two independent determinations were expressed as the area under the curve (AUC) of the aggregation tracing. AUC is reported herein in units (U), as described previously.23
Statistical analysis
Data are expressed as mean±SD. Statistical comparisons were performed with the Mann Whitney U test, the paired and unpaired Student t test and χ2 test. COX regression analysis was performed to compare event rates between the non-HPR group and the individualised treatment group. As the power of the study was limited due to the low event rate, we provide crude and adjusted HR. The adjustment was done for gender, body mass index, diabetes, hyperlipidaemia, use of calcium channel blockers (CCB) and proton pump inhibitors (PPI), clinical presentation, platelet count and cardiogenic shock. All statistical calculations were performed using commercially available statistics analysis software (SPSS V.21; Chicago, USA).
Sample size
We estimated that the sample size of 1008 patients would provide 80% power to demonstrate a reduction in the incidence of ST by individualisation of antiplatelet therapy, on the basis of assumptions of ST rates during 1 month follow-up. We expected a 0.2% rate of ST at 1 month in patients without HPR, as compared to a 1.9% rate in a historical group of patients with HPR.3 ,5 ,14 Thus, if the HR for ST was threefold to fourfold lower in patients without HPR than in those with HPR,3 the study would have more than 80% power to demonstrate that individualised antiplatelet therapy in patients with HPR reduces the rate of ST.
Results
Patient inclusion and baseline characteristics
Of 1043 consecutive PCI patients, only those with unsuccessful reopening of a chronic total occlusion or with conventional balloon-only PCI were excluded (n=35), leaving 1008 participants (figure 2). All STEMI patients received primary PCI. At 30 days, 1 patient (0.09%), a French tourist, was lost to follow-up. Table 1 shows the demographic variables of our patient cohort and differences between the group without HPR after clopidogrel loading (non-HPR) and the individualised group (ie, ADP receptor blocker reloading and primary prasugrel or ticagrelor loading).
Baseline characteristics

Flow chart of study patients. CTO, chronic total occlusion; PCI, percutaneous coronary intervention.
Patients in the individualised group were more frequently of female gender (p=0.01), had higher bodyweight (p=0.001), and a greater incidence of diabetes (p=0.003), especially insulin dependent (p=0.001), STEMI and cardiogenic shock (p<0.001). Higher platelet counts (p<0.001), and co-medication with PPI (p<0.001) and CCB (p=0.03), were also significantly associated with individualisation of DAPT.
Angiographic and interventional details
Table 2 shows angiographic and procedural characteristics according to platelet inhibition (non-HPR vs individualised group).
Angiographic and interventional details
The rate of DES implantation was high (94%), and of these 20% were biolimus-eluting, 49% everolimus-eluting and 25% zotarolimus-eluting. Multivessel disease was present in 65% of patients, with a high proportion of complex lesion morphology (type b2/c: 73%), including 11% left main and 58% left anterior descending artery lesions, resulting in 2.2±1.5 implanted stents/patient (mean stent length 43±33 mm). The rate of use of a femoral access site for PCI during the registry period was high (86%). All parameters showed no differences between groups.
Primary ADP receptor blocker loading and individualisation of ADP receptor blocker therapy
As shown in figure 3A, 94.8% of patients were primarily loaded with 600 mg clopidogrel, 5% with 60 mg prasugrel (STEMI patients <75 years and >60 kg without history of stroke) and 0.2% with 180 mg ticagrelor (known clopidogrel allergy). Of the clopidogrel loaded patients, 30% showed HPR. Clopidogrel reloadings of 600 mg were performed up to three times in 27% of patients with HPR, leaving five patients with persisting HPR, of whom three were finally switched to prasugrel during the observation period, as it became available. Prasugrel reloading was performed in 70% of patients with HPR. Of the prasugrel loaded patients, 2% showed HPR, which was successfully treated with ticagrelor reloading; this was also performed in 3% of patients with HPR to clopidogrel and contraindications to prasugrel. Only three patients remained in HPR during the observation period; they were put on a higher MD (two on clopidogrel 150 mg, one on prasugrel 20 mg as ticagrelor was not yet available). For patients older than 75 years or weighing less than 60 kg, prasugrel 5 mg was primarily prescribed (15% of prasugrel patients, n=37). After MEA testing 1 week later, 14% (n=5) were switched to 10 mg.

Flow chart of primary ADP receptor blocker and acetylic salicylic acid loading and reloading. (A) ADP receptor blocker loading. Only 0.3% of patients (n=3) showed persisting high on-treatment platelet reactivity (HPR) to ADP (≥50 U): two patients after 4×600 mg clopidogrel loading (as prasugrel was not yet available) and one patient on prasugrel (as ticagrelor was not yet available). (B) HPR to AA-induced aggregation (>35 U) occurred to a significant higher proportion in patients with HPR to ADP (ADP ≥50 U). In patients with HPR to ADP and intermediate HPR to AA (AA <60 U) only ADP receptor blocker reloading successfully treated HPR to AA as well. AA, arachidonic acid, ASA, acetylic salicylic acid.
ASA-dependent platelet aggregation and reloading
After ASA and ADP receptor blocker loading, 9% of our patients showed a HPR to AA-induced aggregation (68±28 U vs 16±8 U; p<0.001). As shown in figure 3B, HPR to AA was significantly more prevalent in patients with HPR to ADP (22% vs 4%; p<0.001). HPR to AA without HPR to ADP (63±29 U) was treated by ASA reloading successfully in all patients (14±6 U; p<0.001). In patients with HPR to ADP, the HPR to AA was influenced by the extent of the residual AA-induced platelet aggregation, as follows. In patients with intermediate HPR to AA (<60 U), only ADP receptor blocker reloading was sufficient to treat HPR to AA as well (from 45±7 U to 15±10 U; p<0.001). In patients with high HPR to AA (≥60 U), an additional ASA reloading was necessary to significantly reduce AA-induced aggregation from 92±21 U to 20±16 U (p<0.001). Six of these patients showed persisting HPR to AA and were discharged on 300 mg ASA.
Platelet aggregation in clopidogrel and prasugrel loaded patients and effect of reloading
ADP-induced aggregation after 600 mg clopidogrel loading was significantly higher in patients with HPR (=non-responder: 73±19 U) than without (=responder: 28±11 U; p<0.001; figure 4A). Reloading effectively treated HPR (22±12 U; p<0.001), except in two patients for whom prasugrel was not yet available. ADP-induced aggregation after 60 mg prasugrel loading was significantly higher in patients with HPR (=non-responder: 82±26 U) than without (=responder: 19±10 U; p<0.001), and was successfully treated with ticagrelor reloading (34±15 U; p=0.02; figure 4B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
ADP-induced aggregation after 600 mg clopidogrel or 60 mg prasugrel loading and effect of reloading. (A) Of clopidogrel loaded patients, 30% showed a high on-treatment platelet reactivity (=non-responder), effectively treated by reloading (except for two patients as prasugrel and ticagrelor had not yet been available). (B) Of prasugrel loaded patients, 2% showed a high on-treatment platelet reactivity (=non-responder), effectively treated by ticagrelor.
GPI treatment
GPI was given to 61% (n=57) of STEMI patients, with an intracoronary abciximab bolus only in 91% (n=52) and an intravenous eptifibatide bolus only in 9% (n=5). NSTEMI patients received a GPI treatment in 11% (n=47) of cases, with an intracoronary abciximab bolus only in 72% (n=34) and an intravenous eptifibatide bolus only in 28% (n=13).
Clinical outcome at 30 days
Table 3 shows the clinical outcome of the overall patient cohort.
30-day clinical outcome
No acute ST occurred within 24 h in the whole patient cohort. Three patients died in cardiogenic shock within 24 h after successful PCI without evidence of ST at autopsy. Only one subacute definite ST, which also accounted for the only myocardial infarction, occurred within 30 days (0.09%). This patient had multivessel PCI for NSTEMI, and developed diarrhoea and Gram negative sepsis. On the seventh day post-PCI, an attempted resuscitation was unsuccessful. Acute thrombosis of the circumflex artery stent was confirmed at autopsy. Two sudden deaths without autopsy occurred after discharge in NSTEMI patients, which have been classified as probable ST according to the ARC criteria. However, both patients also suffered from ischaemic cardiomyopathy, which would suggest a primary rhythmogenic cause for their sudden deaths. MACE number equals cardiovascular deaths (n=18; 1.8%) as all three cases of ST died. Cardiogenic shock was the cause of cardiovascular deaths in the majority of cases (88%), without differences in groups. Concerning bleeding complications, no increase in individualised patients occurred (2.6% TIMI major and minor bleedings in both groups). Slightly more than half of the bleeding complications (54%, n=14) were related to the access site (‘instrumented’), requiring surgical intervention in three cases (21% of instrumented complications; 0.3% of patients). The majority of spontaneous bleeding complications were gastrointestinal (67%, n=8). One intracranial haemorrhage occurred under standard DAPT with clopidogrel 17 days after PCI for NSTEMI in an 86-year-old patient.
Table 4 shows 30-day outcomes for the STEMI, NSTE-ACS and stable CAD cohorts.
Descriptive Statistics for 30 days outcome in clinical subgroups
No ischaemic event occurred either in the STEMI cohort, with a required high rate of individualisation (67%), or in the stable CAD cohort, with a sufficient lower rate of individualisation (30%). The safety end point of combined TIMI major and minor bleeding risk was 2× higher in patients with non-ST-elevation ACS (NSTE-ACS) and 4× higher in STEMI patients than in stable patients with CAD (2.9% vs 6.5% vs 1.5%; p=0.02), without an increase associated with individualisation in any subgroup.
Discussion
The main findings of our study are as follows. First, routine efficient peri-interventional individualisation of DAPT with MEA, incorporating the newer generations of ADP receptor blocker (prasugrel and ticagrelor), is able to minimise early ischaemic events after PCI in an all-comers population including STEMI patients by nearly abolishing early definite ST. Second, intensifying platelet inhibition in patients with HPR does not increase bleeding complications compared to patients without HPR under DAPT. Third, there is indirect evidence for synergistic roles of ADP-dependent and ASA-dependent platelet activation.
For interpretation of the very low ischaemic complication rate observed during the 30 days after PCI, the most recent literature on the incidence of real world early ST in PCI for all-comers24 and STEMI patients,25 ,26 as well as the complication rate in the randomised CHAMPION Phoenix trial,11 should be considered. We could show that adjusting the level of platelet inhibition reduced the rate of early definite ST to 0.09%, which is about sevenfold lower than observed in PCI for all-comers24 and about 25-fold to 35-fold lower than in primary PCI for STEMI,25 ,26 even with contemporary second generation DES. Monitored intensification of platelet inhibition by bolus-only administration of GPI and individualised DAPT resulted in a yet more favourable outcome in our STEMI population, as no early thrombotic events occurred. Furthermore, even under randomised study conditions such as the CHAMPION Phoenix trial,11 the definite ST rate after clopidogrel loading was 1.4% within 48 h, or about 14-fold higher than in our study. Immediate ADP receptor blockade with cangrelor, however, showed a benefit with reduction to 0.8% (p=0.01), which is still about eightfold higher than what was achieved with our individualisation protocol. In addition, ischaemic complications were not only not driven by urgent patients with ACS (4.1%), but were also numerically higher in stable CAD (7.4%). By contrast, individualisation of DAPT in our stable CAD cohort, with 600 mg clopidogrel loading the day before PCI and MEA guided individualisation (the latest within 2 h after PCI), resulted in no early ischaemic events.
Three randomised multicentre trials7–9 failed to show a clinical benefit of individualising DAPT with the VerifyNow assay. Among the most common raised limitations, those in study design, protocol implementation and efficacy of platelet inhibition are the most important. Concerning study design, the late randomisation of patients (more than 12 h after PCI) in GRAVITAS7 and TRIGGER-PCI9 excluded acute procedural complications attributable to insufficient platelet inhibition. This occurred even in stable patients with CAD, as is impressively shown in CHAMPION Phoenix.11 Concerning protocol implementation, the ARCTIC trial8 discharged 1.3% of patients in the active study arm without any ADP receptor blocker medication, and lost nearly 9% of patients to follow-up. TRIGGER-PCI9 was stopped prematurely, leaving an underpowered study population. Concerning efficacy of platelet inhibition, 40% of patients in GRAVITAS7 and 16% in ARCTIC8 remained in HPR due to primary reloading with clopidogrel (100% in GRAVITAS and 90% in ARCTIC). By contrast, 100% of our patients were included prior to PCI and discharged with DAPT, 99.9% could be followed up at 30 days and only 0.3% remained in HPR. Together, this resulted in a 1.7-fold lower rate of ST (definite and probable) than in the high-dose clopidogrel arm of GRAVITAS7 and a 3.5-fold lower rate than in the monitored arm of ARCTIC,8 despite our higher risk population, including STEMI patients.
Concerning bleeding complications, our concept of using the newer generations of ADP receptor blockers, primarily for intensifying platelet inhibition in patients with HPR to clopidogrel rather than upfront for all patients with ACS without contraindications, seems beneficial. In contrast to TRITON18 and PLATO,19 which featured significantly increased non-CABG related bleeding rates under prasugrel and ticagrelor, no increased bleeding occurred in the individualised patients compared to those on clopidogrel without HPR. The observed 1.5% TIMI major bleeding rate in our ACS cohort compares favourably to the non-CABG related TIMI major bleeding rates in the clopidogrel arms of TRITON (1.8%) and PLATO (2.2%). Furthermore, even in the highest bleeding risk group, the STEMI patients, our blocking and bridging strategy with GPI bolus-only administration resulted in fewer TIMI major and minor bleeds (6.4%) than in the GPI arm with bolus and infusion (9.6%) of the HORIZON AMI trial.27 Although our number of patients is admittedly far too low to draw this conclusion, GPI bolus-only administration seems suggestively comparable to the bivalirudin arm (5.9%).
Concerning the regulation of platelet activation, it is already known that thrombin-mediated (via the protease activated receptor-1) and ADP-mediated (via the P2Y12 receptor) platelet activation play a synergistic role in haemostasis and thrombosis.20 ,28 ,29 We provide indirect evidence for a synergistic role of ADP-dependent and ASA-dependent (cyclo-oxygenase) platelet activation. We observed an interplay between AA-induced and ADP-induced platelet aggregability, as HPR to AA was significantly associated with HPR to ADP, and solitary reloading with ADP receptor blocker in patients with HPR to ADP and AA was able to successfully resolve intermediate levels of HPR to AA without ASA reloading.
Limitations of our study include primarily the non-randomised nature of the registry without a control group concerning efficacy, and the monocentric design, leading to the need for a high number of indirect comparisons, with all its known shortcomings, in order to discuss and put our findings in perspective.
In conclusion, our data strongly suggest that HPR represents a modifiable risk factor that can be used for tailoring treatment in PCI patients, rather than a marker of higher risk only. Effective individualisation of DAPT for PCI under MEA guidance is able to minimise early ischaemic complications to a so far unreported degree. Further properly designed randomised multicenter trials utilising MEA seem warranted.
Acknowledgments
The authors thank Carina Gruber and Julia Macura for data acquisition, and Doris Langenau and Gabriela Zimmerman for their technical laboratory support. The authors furthermore thank Erich Budschedl, Thomas Chatsakos, Veronika Franke, Fritz Freihoff, Thomas Hafner, Sabine Hoffmann, Susanne Reiter, Edgar Schmidt and Eva Wilhelm, who provided and cared for study patients.
References
Footnotes
-
Contributors GC was involved in the conception and design of the registry, acquisition, analysis and interpretation of data, drafting and revising the manuscript critically for important intellectual content; and final approval of the version to be published; JMS-M was involved in the design of the registry, analysis and interpretation of data, revising the manuscript critically for important intellectual content; and final approval of the version to be published; MF was involved in the design of the registry, acquisition and analysis of data, revising the manuscript critically for important intellectual content; and final approval of the version to be published; CD was involved in the design of the registry, acquisition and analysis of data, revising the manuscript critically for important intellectual content; and final approval of the version to be published; KG was involved in the design of the registry, analysis of data, revising the manuscript critically for important intellectual content; and final approval of the version to be published; AP-S was involved in the design of the registry, interpretation of data, revising the manuscript critically for important intellectual content; and final approval of the version to be published. All authors agreed to be accountable for all aspects of the work and to ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
-
Funding Kaiser Franz Josef Hospital.
-
Competing interests None.
-
Ethics approval The local Ethics Committee of the City of Vienna approved the study protocol in accordance with the Declaration of Helsinki. Participants were included between November 2008 and June 2012.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement No additional data are available.