Article Text

Abstract
Introduction Pertrochanteric hip fractures occur in an elderly population and cause considerable morbidity and loss of functional ability as the fracture heals. Recently, parathyroid hormone (PTH), which is licensed for the treatment of osteoporosis, has been shown to potentially accelerate bone healing in animal and human studies. If its administration could allow a faster functional recovery after pertrochanteric hip fracture, then a patient's hospital stay may be reduced and rehabilitation could be potentially accelerated. PTH can currently only be administered by subcutaneous injection. The acceptability of this intervention is unknown in this elderly population. The aim of this pilot study is to inform the design of a future powered study comparing the functional recovery after pertrochanteric hip fracture in patients undergoing standard care versus those who undergo administration of subcutaneous injection of PTH.
Methods and analysis The study is an open label, prospective, randomised, comparative pilot study with blinded outcomes assessment to establish feasibility of the trial design. Patients will be randomised to receive a 6-week course of PTH or usual treatment. Functional outcomes will be assessed at 6 weeks and 12 weeks. Blinded assessment will be used to minimise the effect of bias of an open label study design. A nested qualitative study will investigate the patient experience of, and expectations following, hip fracture and the patient important aspects of recovery compared with the outcome measures proposed.
Results Results will be analysed to establish the potential recruitment, compliance and retention rates using 95% CIs, and trial outcomes quoted with SDs and 95% CIs for the effect size.
Ethics and dissemination The study has been approved by the South West 2 Research Ethics committee (reference 10/H0206/34). The findings of this study will be disseminated to the medical community via presentations to orthopaedic, orthogeriatric and osteoporosis societies, and their relevant specialist journals.
Trial Registration ISRCTN Register reference number: ISRCTN03362357.
Eudract Number: 2010-020081-22
- Geriatric Medicine
- Rehabilitation Medicine
- Qualitative Research
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Strengths and limitations of this study
-
Blinded assessments.
-
Multicentre, randomised controlled trial.
-
Validated outcome measures used.
-
Qualitative interview of patient experience.
-
Open label study.
-
Pilot study.
-
Multiple questions.
-
Vulnerable elderly population.
Introduction
In the UK, an estimated 70 000 people are admitted to hospital per year due to a hip fracture. The recovery from hip fractures, in terms of outcome for the patient and timescales, requires extensive resources from the health and social services and has great implications on the quality of life and social support for the individual, despite the current treatment options available.1 Patients in this population rarely recover their preinjury mobility and independence.2 ,3
Parathyroid hormone (PTH) is licensed for the treatment of osteoporosis but there is a growing number of animal studies suggesting it can aid fracture healing.4 ,5 What is currently unknown is the effectiveness of this for patients who have sustained hip fractures. A short-term medical addition of PTH to the current management of these patients has the potential to improve the rate of functional recovery and as a consequence to reduce the risks of longer hospital stays, period of dependence on services and potentially quicken functional recovery.
PTH has been shown to improve bone mineral density and reduce risk of refracture in osteoporotic humans.6 Preclinical studies have shown PTH to have a beneficial effect on callus formation, in terms of quality, with regard to trabecular formation, and acceleration of the remodelling phase.7 ,8 The proposed mechanism for these effects is thought to be via increasing osteoblast-induced bone formation and proliferation of mesenchymal cells when mediated by insulin-like growth factor 1 (IGF-1). IGF-1 is thought to be stimulated by the inflammatory mediators following a fracture.9 Therefore, PTH influences the amount of bone laid down, and the rate at which the bone is remodelled, achieving an increased amount of bone tissue, including increases in trabecular thickness.7 This is thought to lead to accelerated healing of fractures. Despite the evidence from animal studies regarding the benefits in treatment of fractures,10–14 the dosage, duration and cost-effectiveness of treatment remain in question.9 Presently, subcutaneous injection remains the only licensed administration route14 and although there are other potential delivery systems under investigation these are not yet licensed for use.15
The number of publications, expert reviews and animal studies discussing the role of intermittent PTH in fracture healing in orthopaedic, gerontology and endocrinology literature demonstrates the high level of interest and belief in this new application of PTH. Currently, the impact that this treatment may have on patients and their recovery is unknown. The use of a daily injection therapy in elderly acutely injured patients can be viewed as difficult to implement. This pilot study is necessary to investigate how reasonable it is to expect this population to be able to cope with the injection therapy and whether, due to the number of unknown circumstances within the study design, including the proposed outcome measures, it will function appropriately for a full-scale, appropriately powered study which will answer the question of efficacy.
Primary objectives
-
To establish the potential recruitment, compliance and retention rates for this intervention and trial design to inform sample size calculation, feasibility and design for the full study.
-
To trial the outcome measures intended for use in the future full study—to test for time and ease of completion in follow-up clinics and tolerance of participants and to explore the validity and appropriateness of the suggested measures from the patients’ perspective.
-
To clarify and define ‘standard’ medicinal care for osteoporotic fractures received by the comparison group.
-
To establish the feasibility and acceptability of injection therapy over 6 weeks in the elderly and acutely injured population.
Methods
This is an open label, prospective, randomised, comparative pilot study with blinded outcomes assessment to trial the study design. Patients will be randomised to receive PTH or normal treatment. A nested qualitative study will investigate the patient experience of, and expectations following, hip fracture and the patient important aspects of recovery compared with the outcome measures proposed (figure 1).

Trial flow diagram.
Sample
Sample size
This study is intended to be a pilot study to determine whether the methodology is appropriate for a main study with adequate power. As such, a suitable sample size for the pilot study was deemed to be 20 per group. Forty patients will be sufficient to provide estimates of SD alongside published literature and 20 patients will be sufficient to estimate compliance levels to inform a larger adequately powered study. This should also provide sufficient participants for the nested qualitative study (a purposive sample of up to 30 participants) to reach saturation of responses16 in semistructured interviews.
Recruitment
Consecutive patients admitted with a pertrochanteric femoral fracture over the age of 60 years will be identified from the orthopaedic trauma units in six UK acute care National Health Service (NHS) hospitals. Exclusion criteria included the contraindications for the use of PTH detailed in the product literature and those fractures not treated by fixation (box 1).
Exclusion criteria
Exclusion
-
Fracture not as a result of a low-energy injury/fall, for example, fall from standing height
-
Patients whose fracture is managed conservatively
-
Surgical fixation with total hip replacement (THR), haemiarthoplasty or cannulated screws
-
Previous treatment with parathyroid hormone (PTH) or other PTH analogues
-
Hypersensitivity to the active substance or to any of the excipients
-
Previous intravenous bisphosphonate (eg, zoledronic acid) in the previous 12 months
-
Strontium therapy for osteoporosis within the past12 months
-
Current medications for breast and prostate cancer (eg, tamoxifen, anastrozole, Zoladex and Prostap) or other hormone therapies such as testosterone and hormone replacement therapy
-
Decreased capacity to understand the risks of participating in the trial
-
Pre-existing metabolic bone disease, for example, Paget's disease and hyperparathyroidism other than primary osteoporosis or glucocorticoid-induced osteoporosis
-
Pre-existing hypercalcaemia or high or low corrected calcium which requires investigation
-
Severe renal failure (epidermal growth factor receptor <30) or urolithiasis
-
Current unexplained raised alkaline phosphatase
-
Active cancer diagnosis or skeletal malignancies or bone metastases, or prior external beam or implant radiation therapy to skeleton within the past 5 years
-
Premenopausal
-
Pregnancy or lactation
-
Sustained use of oral steroids
-
Wheelchair users, bed bound or transferring only prior to fracture
-
Other current injuries (including fractures) that will affect ability to mobilise at 6 weeks
-
Physically incapable to carry out treatment protocol or appropriate social circumstances (eg, needle phobia, other severe disabilities limiting manipulation of injection pen and without appropriate carer willing and able to assist).
-
Patient consents to study >7 days postsurgery
-
Current participation in any other clinical trial of medicinal product
Appropriate patients will be approached regarding the study as per International Conference on Harmonisation–Good Clinical Practice (ICH–GCP) guidelines. They will be provided with a written patient information sheet and the trial will be discussed, including the concept of randomisation and an explanation of the treatment and follow-up included in both arms of the trial (including potential side effects of the intervention and potential participation in the qualitative substudy). The patient will be given a minimum of 24 h to discuss their participation with whomever they choose. They will also be given contact information of the research team during this time for any further information required. Where possible, potential patients will be given as long as required to consider their participation. However, consent will need to be gained and randomisation performed within 7 days of surgery, permitting initiation of the intervention within 10 days postsurgery. Randomisation will be performed using the secure online service provided by Sealed Envelope (http://www.sealedenvelope.com).
Interventions
PTH intervention arm
The intervention arm will administer a subcutaneous injection of 20 μg recombinant PTH (teriparatide, Eli Lilly, Indiana, USA) from a prefilled pen daily for 6 weeks (42 days). The current research suggests a treatment regime of daily subcutaneous injection, between 21 and 56 days,13 ,17 ,18 which closely matches the requirement for subcutaneous injections of low molecular weight heparin for thromboprohylaxis advised for this patient group.19 As a consequence, a 6-week period of treatment was decided for this study due to matching the current requirements for subcutaneous injections for thromboprophylaxis. Additionally, if 6 weeks do not give a significant benefit, then the clinical improvement anticipated is unlikely to lead to functional improvement of the patient.
A recent study completed by Eli Lilly18 looked at the effect of intermittent PTH on distal radial fracture healing. Measuring the time to radiographic healing, the results showed a reduced healing time in the 20 µg/day treatment group. The results did not show a significant reduction in time to healing measured by cortical bridging on radiograph for the 40 µg/day group compared with the placebo group and no dose response was observed between the 20 and 40 µg/day groups (given for 8 weeks). This supports the intention to trial 20 µg/day. A greater functional difference should be demonstrable in hip fracture healing compared with the upper limb healing due to the role in weight-bearing, essential in activities of daily living. There may be a greater demonstrable effect in the hip fracture population if the extent of osteoporosis is greater. It is recognised by the study group that there is discussion regarding the potential reduction of effect of PTH if there is vitamin D deficiency20 and this will therefore be monitored in all patients.
Prescription of calcium and vitamin D will continue as per standard treatment. Prescription of other osteoporosis medications such as bisphosphonates and strontium will be stopped or delayed until following the 6-week PTH treatment time. Teriparatide is licensed for the treatment of severe osteoporosis and the prevention of further fragility fracture; therefore, delaying the initiation of bisphosphonate therapy does not present any risk to the patients. A review of osteoporosis medication would be indicated as standard care if a patient has been receiving a bone protection medication and has suffered another fragility fracture. After completion of the 6 weeks PTH treatment time, the patient may begin or continue normal osteoporosis treatment which will be prompted by a letter to the participant's general practitioner (GP).
The participants randomised to the intervention arm will be assisted with the injection in the immediate postoperative phase. They will receive training to administer the injection individually from the clinical or research team prior to discharge and provided with written supporting information. The option of a carer administering the injection will be discussed as appropriate when the situation occurs.
Participants will be asked to return the injection pens to the research team at the 6-week follow-up. This will enable a calculation of compliance to be performed.
Normal treatment arm
The normal treatment arm will continue with standard treatment regimens including continuation of or initiation of osteoporosis investigations/medications as per the National Institute for Health and Care Excellence (NICE) guidelines (figure 2).21

The National Institute for Health and Care (NICE) guidelines for osteoporosis treatment.21
Participants in the pilot study will continue to be referred for Dual-energy X-ray Absorptiometry (DEXA) scans in line with normal care standards, for example, as stipulated above in the NICE guidelines21 or if further investigation is clinically indicated. Both arms of the study will undergo operative fixation, postoperative rehabilitation and discharge planning as provided as standard care.
Data collection
Details regarding eligibility will be recorded during the screening process. Further information regarding questions and concerns posed by those approached will be retained to inform the recruitment information for the full trial and the content of the patient information for the full trial as part of the primary objective. Numbers of patients fulfilling the inclusion criteria but not recruited, and where possible, reasons for this will be recorded and monitored to ensure against selective entry and to inform the recruitment process and estimates for the future full trial. Recruitment estimates in the full trial will be based on the recruitment rate from the pilot study in combination with data records of the total eligible patients in each site.
Baseline data (including prefracture social circumstances, concomitant illnesses and medications) will be collected following participant recruitment to allow comparisons between groups. The patients’ health records/attendance to hospital (and primary care records if required) will be monitored up to 1 year for incidence of further fracture and mortality, results of DEXA scan and initiation of osteoporosis treatment.
Outcome measures
The Short Physical Performance Battery (SPPB),22 36 item, Short Form health survey, V.1 (SF-36), EuroQol (EQ-5D) and visual analogue scale (VAS) for pain on weight-bearing measures will be performed by a blinded trained assessor during outpatient clinic visits at 6 weeks and 3 months postoperation. The assessor will be trained by the trial coordinator to ensure consistency and adherence to the trial protocol. Radiograph and compliance information will also be completed as part of their consultation. Patients participating in the nested qualitative study will be asked to repeat all of these assessments during their interviews at 6 and 12 months. Those not participating in the interviews will receive a telephone call to complete the quality of life questionnaires (SF-36 and EQ-5D) at 6 months (figure 3).

{kind=link}
{kind=link}
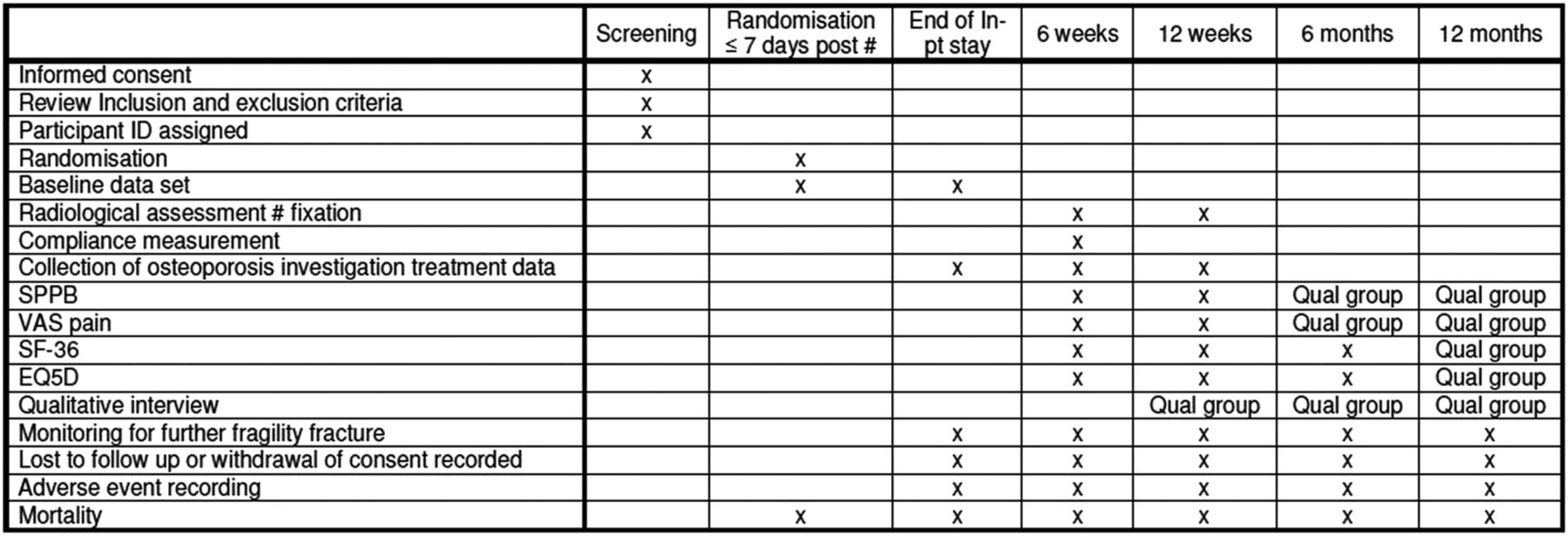{kind=link}
Participant timeline.
Compliance
Patients deviating from any prescribed intervention (intervention or comparative care group) will be encouraged to discuss this with the research team at the 6-week and 3-month follow-ups. The patients in the intervention group will be asked to return their injection pens to the research team at the 6-week follow-up. The remaining contents of the returned pens will be measured, enabling an assessment of compliance.
Monitoring adherence to prescribed medications will continue for the comparison and intervention groups until the 12-week follow-up to allow an improved definition of ‘normal treatment’ for the full study. This will include recording which medications, time of starting medication, timing and results of DEXA scan and tolerance and compliance with their treatment at each follow-up stage.
Participants will be made aware throughout that they may withdraw from the study at any stage. In the occurrence of withdrawal from the study, the patients will be asked if they would inform the research team why they are withdrawing—this is due to the need to establish acceptability of treatment. This would be included in the patient information sheet, and patients would be made aware from the outset. Patients withdrawing/not complying with the injection therapy will be asked if they would continue to participate in follow-up for outcome data collection. Patients withdrawing from attending for follow-up, unless explicitly withdrawing consent, will continue to be monitored for DEXA scan results, further fragility fractures and mortality for 1 year (via hospital or primary care records).
Any participant withdrawing from the study who is part of the qualitative group would be asked if they wished to continue to participate in the semistructured interviews. This is necessary to establish a true picture of acceptability and success of outcome measures and study design. Patients will be made aware that it is their choice whether to take part at each stage.
Safety reporting and monitoring
Participants will be asked to report adverse events (AEs) at each follow-up. The research team will inform the participant's GP of the person's inclusion in the trial on discharge from hospital and encourage timely communication of any AEs experienced during the study. All AEs will be recorded in the study paperwork. The trial coordinator will maintain a log of AEs and inform the chief investigator (CI). All AEs and adverse reactions for both groups will be collated in a document to enable comparison. The CI and trial coordinator will jointly collate reports for submission to the sponsor, Medicines and Healthcare Regulatory Agency (MHRA) and Research Ethics Committee (REC) as required.
All serious adverse events and serious adverse reactions will be reported to the Sponsor via the Research & Development office and the regulatory authorities as per the North Bristol NHS Trust standard operating procedure as soon as the research team is aware of it (within 24 h) and a written report will follow within 7 days as per ICH–GCP guidelines.
The study will be monitored by the North Bristol NHS Trust Research & Innovation office as the sponsor of the study according to their standard operating procedures.
Blinding
The study will be open label due to the complexities of placebo injections in a frail elderly population (the invasive nature of the intervention and the need to withhold or account for the normal osteoporosis medications for the placebo group). This is the plan for the full study. Blinded assessment of an objective functional score has been included to minimise the bias effect this design may have.
Data management
All data collection forms are anonymised. The coordinating centre will check for any missing data or anomalies that can be addressed by the recruiting site. All data will be coded and manually entered into a Microsoft Access 2003 database. Data validation will occur in 10% of all data entry to minimise transcription error. In the event that there is a >2% error overall, the validation will be extended to 20%.
Analysis
Analysis of the feasibility aspects of the pilot study will focus on proportion of patients who were recruited and compliant using 95% CIs calculated using the binomial method.
Statistical analysis for the trial outcomes will be similar to that for the main study, but reporting SDs of outcome variables and CIs of effect sizes, not investigating statistical significance. The two groups will be compared by tabulating information available at baseline (prior to randomisation). Comparisons of outcome variables following randomisation will be carried out using an intention-to-treat methodology and an appropriate methodology will be used where follow-up outcome data are not available. In the full study, this may mean data imputation using regression techniques, but in the pilot study, there will be insufficient data to carry out the regressions robustly and last observation carried forward may be used to prevent omission of patients. Treatment effects between the groups will be calculated and CIs calculated. In the full study, in the unlikely event of large differences between the groups at baseline, regression techniques may be employed to demonstrate the treatment effect before and after controlling for these differences.
Thematic analysis of the qualitative data will be undertaken.16 Interview transcripts will be coded and themes produced from those codes will be used to recommend domains which describe the process of ‘recovery’ or ‘getting better’ from the patients’ perspective and priorities.
The interviews will be audio-recorded and fully transcribed to allow coding and analysis in an ongoing and iterative manner.23 Interview transcripts will be anonymised using the participants’ trial ID. The researcher will code the transcripts according to themes that emerge from the data with supervision from the university supervisors. Data management will be assisted using NVivo software.
Ethics and dissemination
We will comply with the Medical Research Council Good Clinical Practice guidelines, and the trial will run under the standard operating procedures of North Bristol NHS Trust. An independent data monitoring committee will meet annually, with an option to increase if specific concerns arise. The findings of this study will be disseminated to the medical community via presentations to orthopaedic, orthogeriatric and osteoporosis societies and their relevant specialist journals. The protocol amendments to date are listed in table 1.
Protocol amendments
Acknowledgments
The authors would like to thank North Bristol National Health Service (NHS) Trust for agreeing to sponsor the study.
References
Footnotes
-
Contributors TC and RF conceived the original study and developed the protocol with RH, SB, RG, KH, KJ, SL and KW. Statistical advice was provided by RG. TC led the writing of the first draft of the manuscript, with contributions from RH, SB and RF. All authors contributed to the editing and redrafting.
-
Funding This is a summary of independent research funded by the National Institute for Health Research (NIHR)'s Research for Patient Benefit Programme (Grant Reference Number PB-PG-0408-16292).
-
Competing interests None.
-
Ethics approval South West 2 Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement No additional data are available.