Article Text

Abstract
Objective To assess whether three novel interventions, formulated based on a systems medicine therapeutic concept, reduced disease activity in patients with relapsing–remitting multiple sclerosis (MS) who were either treated or not with disease-modifying treatment.
Design A 30-month randomised, double-blind, placebo-controlled, parallel design, phase II proof-of-concept clinical study.
Settings Cyprus Institute of Neurology and Genetics.
Participants 80 participants were randomised into four groups of 20 each. A total of 41 (51%) patients completed the 30-month trial. The eligibility criteria were an age of 18–65; a diagnosis of relapsing–remitting MS according to the McDonald criteria; a score of 0.0–5.5 on the Expanded Disability Status Scale (EDSS); MRI showing lesions consistent with MS; at least one documented clinical relapse and either receiving or not a disease-modifying treatment within the 24-month period before enrolment in the study. Patients were excluded because of a recent (<30 days) relapse, prior immunosuppressant or monoclonal antibody therapy, pregnancy or nursing, other severe disease compromising organ function, progressive MS, history of recent drug or alcohol abuse, use of any additional food supplements, vitamins or any form of polyunsaturated fatty acids, and a history of severe allergic or anaphylactic reactions or known specific nutritional hypersensitivity.
Interventions The first intervention (A) was composed of Ω-3 and Ω-6 polyunsaturated fatty acids at 1:1 wt/wt. Specifically, the Ω-3 fatty acids were docosahexaenoic acid and eicosapentaenoic acid at 3:1 wt/wt, and the Ω-6 fatty acids were linoleic acid and γ-linolenic acid at 2:1 wt/wt. This intervention also included minor quantities of other specific polyunsaturated, monounsaturated and saturated fatty acids as well as vitamin A and vitamin E (α-tocopherol). The second intervention (B, PLP10) was a combination of A and γ-tocopherol. The third intervention (C) was γ-tocopherol alone. The fourth group of 20 participants received placebo. The interventions were administered per os (by mouth) once daily, 30 min before dinner for 30 months.
Main outcome measures The primary end point was the annualised relapse rate (ARR) of the three interventions versus the placebo at 2 years. The secondary end point was the time to confirmed disability progression at 2 years.
Results A total of 41 (51%) patients completed the 30-month trial. Overall, for the per-protocol analysis of the 2-year primary end point, eight relapses were recorded in the PLP10 group (n=10; 0.40 ARR) versus 25 relapses in the placebo group (n=12; 1.04 ARR), representing a 64% adjusted relative rate reduction for the PLP10 group (RRR 0.36, 95% CI 0.15 to 0.87, p=0.024). In a subgroup analysis that excluded patients on monoclonal antibody (natalizumab) treatment, the observed adjusted RRR became stronger (72%) over the 2 years (RRR 0.28, 95% CI 0.10 to 0.79, p=0.016). The per-protocol analysis for the secondary outcome at 2 years, the time to disability progression, was significantly longer only for PLP10. The cumulative probability of disability progression at 2 years was 10% in the PLP10 group and 58% in the placebo group (unadjusted log-rank p=0.019). In a subgroup analysis that excluded patients on natalizumab, the cumulative probability of progression was 10% for the 10 patients in the PLP10 group and 70% for the 12 patients in the placebo group, representing a relative 86% decrease in the risk of the sustained progression of disability in the PLP10 group (unadjusted log-rank p=0.006; adjusted HR, 0.11; 95% CI 0.01 to 0.97, p=0.047). No adverse events were reported. Interventions A (10 patients) and C (9 patients) showed no significant efficacy.
Conclusions In this small proof-of-concept, randomised, double-blind clinical trial; the PLP10 treatment significantly reduced the ARR and the risk of sustained disability progression without any reported serious adverse events. Larger studies are needed to further assess the safety and efficacy of PLP10.
Trial registration International Standard Randomised Controlled Trial, number ISRCTN87818535.
- Nutrition & Dietetics
- Complementary Medicine
- Public Health
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Article summary
Article focus
-
Increasing prevalence of multiple sclerosis (MS), combined with the limited efficacy and side effects of existing treatments, urges the development of new, innovative, more effective, and safe, preventive treatment strategies.
-
We propose three novel nutraceutical treatment interventions, formulated on a systems medicine rationale through nutritional systems biology, with PLP10 representing the complete composition of the formulation.
-
We studied the proposed interventions in a randomised, double-blind, placebo-controlled, phase II proof-of-concept clinical trial.
Key messages
-
In this small trial, the PLP10 treatment statistically significantly reduced the ARR and the risk of sustained disability progression without any reported serious adverse events.
-
A total of 41 (51%) patients completed the 30-month trial. For the per-protocol analysis of the primary end point, we observed a 64% relative rate reduction for the PLP10 group. The cumulative probability of disability progression at 2 years was 10% in the PLP10 group and 58% in the placebo group.
Strengths and limitations of this study
-
▪ Randomisation, blinding, use of placebo, definite inclusion/exclusion criteria and primary/secondary end points, along with the extended study period, allowed for an appropriate overview of safety and efficacy.
-
▪ The small sample size and the high rate of dropouts are limitations associated with the study.
Introduction
Multiple sclerosis (MS) is a complex multifactorial disease that results from the interplay between environmental factors and a susceptible genetic background.1–3 Together, these factors trigger a cascade of events involving the engagement of the immune system, inflammatory injury of myelin, axons and glia, functional recovery and structural repair, gliosis and neurodegeneration.4 The mechanisms involved include immune-mediated inflammation, oxidative stress and excitotoxicity, all of which contribute to oligodendrocyte and neuronal damage and even cell death, hence promoting disease progression.5–9 The increasing prevalence of MS, combined with the partial efficacy and side effects of the existing treatments, have urged the development of new, innovative, more effective, safe and preventive treatment strategies.
Recent research has shown that multiple variables dynamically interact and many different complex inter-related processes are simultaneously orchestrated for MS pathogenesis. The uniqueness of systems medicine (SM) is the recognition that different specific complex factors are important in disease management and that these factors need to be incorporated in some meaningful way for treatment selection and delivery.10 The primary challenge of a systems scientific approach is the elucidation of how these multiple variables dynamically interact and how this understanding can be applied to affect the system and achieve a desirable end.10 ,11 One approach towards that end might be the simultaneous intervention in multiple involved pathways using a combination of different active ingredients that could exert a synergistic effect and provide a comprehensive, sustainable treatment effect (see online supplementary information methods 1).
The polyunsaturated fatty acid (PUFA) composition of membrane phospholipids plays an important role in immune-related and non-immune-related inflammation. PUFA and antioxidant deficiencies, along with decreased cellular antioxidant defence mechanisms, have been reported in MS patients.12–15 The cause of PUFA deficiencies is not entirely clear and may involve metabolic and nutritional alterations.12
Increased or uncontrolled inflammation contributes to several different acute and chronic diseases, and it is characterised by the production of inflammatory cytokines, arachidonic acid (AA)-derived eicosanoids (prostaglandins (PGs), thromboxanes (TXs), leukotrienes (LTs) and other oxidised derivatives), and other inflammatory agents such as reactive oxygen species (ROS), nitric oxide (NO) and adhesion molecules (figure 1).16 During inflammation, glutamate homeostasis is altered by the release of increased quantities of glutamate by activated immune cells, which can result in the overactivation of glutamate receptors and, in turn, excitotoxic oligodendroglial death.7 ,17 Among others, membrane-related pathology, immune-mediated inflammation, oxidative stress and excitotoxicity provide potentially useful combined targets for intervention in MS.
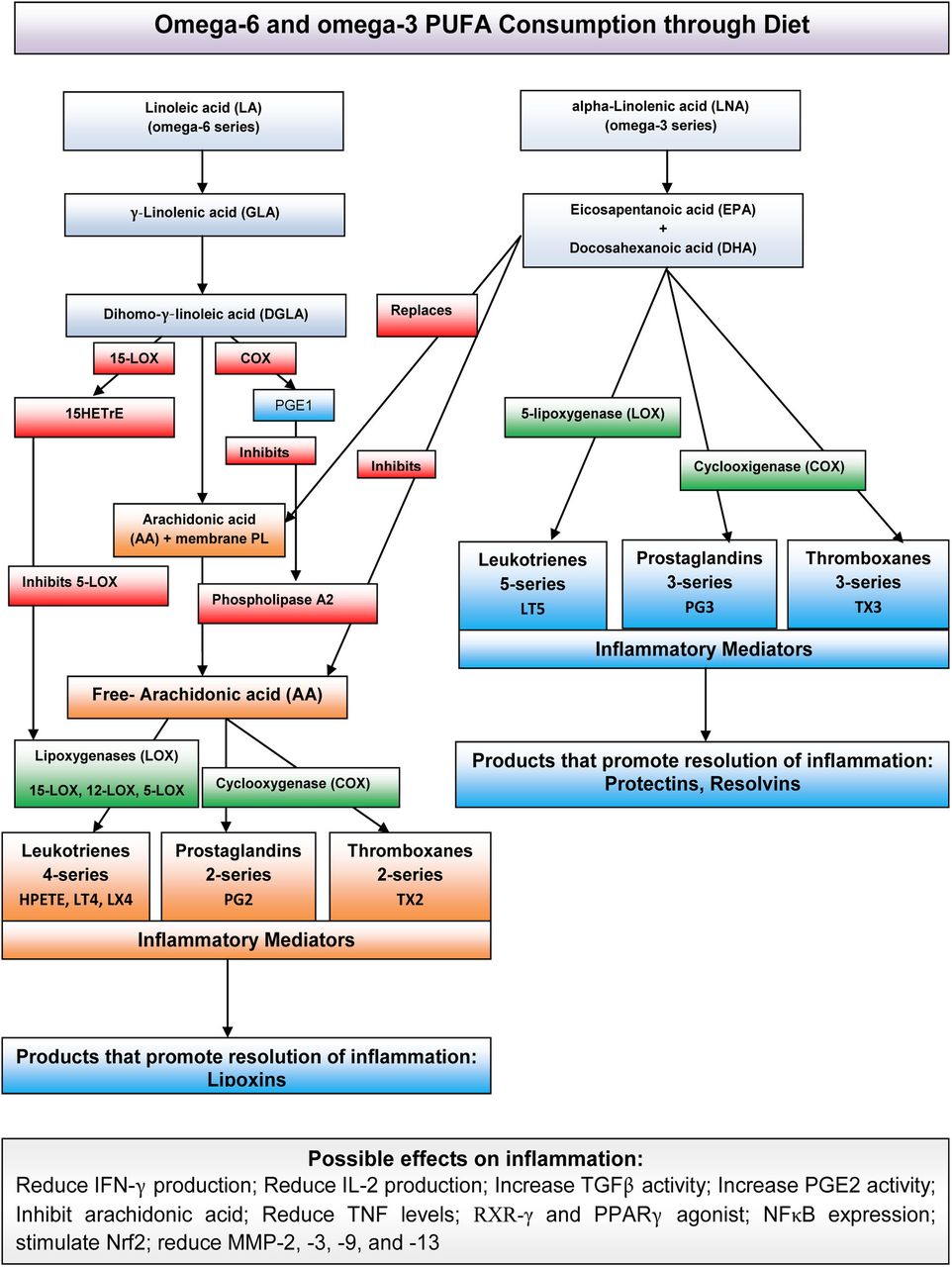
Ω-6 and Ω-3 PUFAs, their respective metabolic derivatives and their possible effects on inflammation. After consumption, the PUFAs are metabolised via several pathways (not shown) to active compounds that mediate inflammation and to products that promote the resolution of inflammation. COX, cyclooxygenase; HETrE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid; IFN-γ, interferon γ; IL-2, interleukin 2; LOX, lipoxygenase; LT, leukotriene; MMP, metalloproteinase; NFκB, nuclear factor kappa B; Nrf2, nuclear respiratory factor; PG, prostaglandin; PGE2, prostaglandin E2; PL, phospholipid; PPARγ, peroxisome proliferator-activated receptor γ; PUFAs, polyunsaturated fatty acids; RXR-γ, retinoid X receptor/γ; TGFβ, transforming growth factor β; TNF, tumour necrosis factor; TX, thromboxane.
In vitro and in vivo studies have demonstrated that dietary eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), linoleic acid (LA) and γ-linolenic acid (GLA) can be implicated and modulate almost all known complex networks of events and pathways in MS pathophysiology. The brain membrane fatty acid composition can be modified with dietary supplementation, but the process has been shown to be age dependent (taking much longer in adults vs developing brains) and possibly dependent on the quantity of the dietary/supplemented PUFAs.18 Both human and animal studies have proved that diets high in DHA and EPA can increase the proportion of these PUFAs in the membranes of inflammatory cells and also reduce the levels of AA, a stress-related biomarker.16 ,19 The anti-inflammatory properties of Ω-3 PUFAs include the production of PGs and TXs of the 3-series and of LTs of the 5-series (figure 1).18 ,20 Resolvins and protectins are biosynthesised from Ω-3 fatty acids via cyclooxygenase-2/lipoxygenase (COX-2/LOX) pathways, and they promote the control of inflammation in neural tissues (figure 1).21–25 T- cell proliferation in acute and chronic inflammation can also be reduced by supplementation with either Ω-6 or Ω-3 PUFAs.26 Furthermore, vitamin E is an important antioxidant that can interrupt the propagation of free radical chain reactions.27 Specifically, vitamin E (α-tocopherol, an isoform of vitamin E) efficiently detoxifies hydroxyl, perhydroxyl and superoxide free radicals, whereas γ-tocopherol (another isoform of vitamin E) appears to be more efficiently implicated in trapping NO radicals.28 ,29 In addition, α-tocopherol exerts non-antioxidant properties, including the modulation of cell signalling and immune functions, regulation of transcription and induction of apoptosis.30
Moreover, Ω-3 fatty acid electrophilic derivatives formed by COX-2 in activated macrophages can stimulate the nuclear respiratory factor (Nrf), which induces the transcription of neuroprotective and antioxidant-related genes and can activate the peroxisome proliferator-activated receptor γ (PPARγ) for an anti-inflammatory response.31–34 In animal studies, EPA and DHA have proved to be endogenous ligands of the retinoid X receptor (RXR), with positive effects on neurogenesis.35 Additionally, in 2008, Salvati et al reported evidence of accelerated myelination in DHA-treated and EPA-treated animals.36 Moreover, DHA and EPA have been reported to significantly decrease the levels of metalloproteinases (MMP)-2, MMP-3, MMP-9 and MMP-13, which have a significant role in the migration of lymphocytes into the central nervous system by inducing the disruption of the blood brain barrier, an important step in the formation of MS lesions.37–43
Based on the aforementioned observations, specific PUFAs and antioxidant vitamins fulfil the criterion of biological plausibility and have the potential to diminish the severity and activity of MS symptoms, potentially even promoting recovery (remyelination).12 ,44
We report here a randomised phase II, single-centre, double-blind, placebo-controlled, proof-of-concept clinical trial evaluating the therapeutic ability of a nutraceutical formula (with PLP10 representing the complete composition of the formulation) and of two other interventions (A and C) consisting of PLP10-constituent partial fractions containing ingredients for the aforementioned substance categories on relapsing remitting MS patients.
Methods
Patients
The eligibility criteria were an age of 18–65; a diagnosis of RRMS according to the McDonald criteria; a score of 0.0–5.5 on the Expanded Disability Status Scale (EDSS), a rating that ranges from 0 to 10, with higher scores indicating more severe disability; MRI showing lesions consistent with MS; at least one documented clinical relapse; and either receiving or not a disease-modifying treatment (DMT) within the 24-month period before enrolment in the study. Patients were excluded because of a recent (<30 days) relapse, prior immunosuppressant or monoclonal antibody therapy, pregnancy or nursing, other severe disease compromising organ function, progressive MS, history of recent drug or alcohol abuse, use of any additional food supplements, vitamins, or any form of PUFA and a history of severe allergic or anaphylactic reactions or known specific nutritional hypersensitivity. No monitoring or limitations on the patients’ daily dietary habits were considered because the high quantities of the ingredients within the formula could not be significantly affected by any particular dietary pattern.
The study was conducted in accordance with the standards of the International Conference of Harmonisation Guidelines for Good Clinical Practice. The protocol was developed by the investigators, approved by the Cyprus National Bioethics Committee, and overseen by an independent safety-monitoring committee evaluating the safety and over-all benefit-risk profiles. The adherence of the care providers with the protocol was assessed by an external committee assigned by the funder of the project through reviews of case report forms. All patients gave written informed consent at the time of enrolment.
Randomisation and masking
Patients were randomly assigned to four intervention groups in a 1:1:1:1 ratio stratified by gender (women to men, 3:1). Randomisation was facilitated by a lottery-type pool of numbered balls. Patients were randomly assigned to the treatments in blocks of four by flipping a coin as follows: for the first two drawn balls, heads stratified them to the groups A/B and tails stratified them to the groups C/D. The other two balls were stratified accordingly. A second toss of the coin assigned the two patients to group A (head)/B (tail) or to group C (head)/D (tail). The randomisation scheme was generated, performed and securely stored by the Helix Incubator Organization of Nicosia University (HIONU).
The interventions had identical appearances and smells and were kept in dark bottles (15 daily-dose portions/bottle) under a nitrogen bed and labelled by HIONU with code numbers, blinded for both the patients and investigators. Study data were collected by the investigators and saved by HIONU, which also held the blinded codes for the study. All study personnel involved in the conduct of the study were blinded throughout the study. The treating/examining physician, other investigators, pharmacist, neuroradiologist and patients were masked to the treatment allocation.
Procedures and end points
The specific Ω-3 and Ω-6 raw materials were purchased according to the required interventions’ PUFA-fraction specification (molecular structure, quantity/ratio and quality) with vitamin E (α-tocopherol) used as antioxidant stabiliser by the supplier. The vitamins and masking aroma were purchased separately. The mixing of the fractions to the final required intervention-composition specification was always performed by the same team of scientists under the supervision of the involved medical biochemist and lipidology specialist and under appropriate conditions every 6 months. The interventions were refrigerated in the dark until use. See table 1 and supplementary information methods 1 and 2 for a detailed description of the interventions.
Intervention ingredients per-treatment arm
The participants were randomly assigned to receive the following: group A, a daily dose of a 19.5 ml mixture of EPA (1650 mg)/DHA (4650 mg)/GLA (2000 mg)/LA (3850 mg)/total other Ω-3 (600 mg)/total monounsaturated fatty acids (MUFA) (1714 mg)+total saturated fatty acids (SFA) (18:0 160 mg and 16:0 650 mg)/vitamin A (0.6 mg)/vitamin E (22 mg) plus citrus-aroma (intervention A); group B (PLP10), a daily dose of a 19.5 ml mixture of EPA (1650 mg)/DHA (4650 mg)/GLA (2000 mg)/LA (3850 mg)/total other Ω-3 (600 mg)/total MUFA (1714 mg)+total SFA (18:0 160 mg and 16:0 650 mg)/vitamin A (0.6 mg)/vitamin E (22 mg) plus pure γ-tocopherol (760 mg) plus citrus-aroma (intervention B); group C, a daily dose of a 19.5 ml mixture of pure γ-tocopherol (760 mg) dispersed in pure virgin olive oil (16,137 mg) plus citrus-aroma (intervention C) and group D (placebo), a daily dose of a 19.5 ml mixture of pure virgin olive oil (16 930 mg) plus citrus-aroma (intervention D; table 1). The pharmacist of the institution was responsible for the appropriate storage and handling of the interventions for the individual participants. The interventions were taken orally once daily 30 min before dinner using a dosage-calibrated cup for 30 months. The ingredients, ratio and dose were selected based on their biophysical interrelationship with the total known multiple MS causative factors, their biochemical importance and the role they were expected to play in the normalisation and treatment of the involved complex network of events in the disease pathophysiology. Moreover, the high intervention dosage was selected with the aim of optimising the body composition of Ω-3 to Ω-6 PUFAs to a 1:1 wt/wt ratio, irrespective of the dietary habits and geographical origin.
The period from 1 July 2007 (enrolment) to 31 December 2007 was used as the normalisation period. This 6-month normalisation period would allow the interventions to exert their beneficial effect as oral PUFAs need 4–6 months to achieve pivotal action on immune and neural cells, correction of antioxidant deficiencies and body PUFA redistribution, and an optimal normalisation of the EPA and DHA ratios.45–47 The study was completed on 31 December 2009 (30 months), and the recording of relapses continued until 31 December 2010 (42 months). Overall, the study included a ‘normalisation period’ (1 July 2007 to 31 December 2007), an ‘on treatment’ period (1 January 2008, the baseline, to 31 December 2009) and a 12-month ‘post-study monitoring period’ (1 January 2010 to 31 December 2010).
Depending on their clinical status and in accordance with common practice, the participants continued to receive their indicated regular treatment, with persistent evaluation for any side effects and adverse events. Clinical assessment visits were scheduled at baseline and at 3, 9, 15, 21 and 24 months on-treatment. The patients were also clinically examined by the treating neurologist within 48 h after the onset of new or recurrent neurological symptoms.
The primary end point was the annualised relapse rate (ARR) at 2 years. A relapse was defined as new or recurrent neurological symptoms not associated with fever or infection that lasted for at least 24 h and was accompanied by new neurological signs. Relapses were treated with methyl-prednisolone at a dose of 1 g intravenous per day for 3 days, followed by prednisone orally at a dose of 1 mg/kg of weight per day on a tapering scheme for 3 weeks. The secondary end point at 2 years was the time to disability progression, defined as an increase of 1.0 or more on the EDSS and confirmed after 6 months. Progression could not be confirmed during a relapse, and the final EDSS score was confirmed 6 months after the end of the study. A post hoc analysis was performed to assess the proportion of patients free from new or enlarging T2 lesions on brain MRI scans at the end of the study for the per-protocol participants of the group receiving the most effective intervention versus placebo. This comparison was made versus the available archival MRI scans up to 3 months before the enrolment date. The MRI scans were performed and blindly analysed at an MRI evaluation centre. The patients were monitored for an additional 12 months after completion of the trial, and relapses were recorded. The patients were strongly encouraged to remain in the study for follow-up assessments even if they had discontinued the study drug.
Blood samples were collected from all randomised patients at the time of enrolment, at every scheduled clinical assessment and during relapses. To evaluate the compliance, the fatty acid composition of the patients’ red blood cell membranes was determined by gas chromatography, according to a standard protocol. The fatty acid analyses were performed after study termination and thus did not influence the blinding. Safety measures were assessed from the time of enrolment until 12 months following the study completion. Haematological and biochemical tests were performed at enrolment and every 12 months, including a full blood count, renal and liver function tests, and protein, cholesterol, triglyceride, glucose and electrolyte levels.
The involved neurologist was experienced with more than 20 years in practice. He was trained to standardise the EDSS scoring procedures, examined the patients, made all medical decisions, determined the EDSS score and reviewed the adverse effects or side effects. The medical biochemist, who was a specialist in lipidology and immunology, and the registered clinical dietician were both members of the investigative team with more than 25 years of experience in practice. The patients were able to contact the involved neurologist at any time if there was any adverse event, side effect or allergic reaction. The study drug was not expected to have any clinical or laboratory adverse effects different from those of the placebo that could disturb the double-blind nature of the trial. Therefore, the study neurologist functioned as both the treating and evaluating physician.
The whole procedure followed the clinical trial guidelines as required by the USA Food and Drug Administration, European Medicines Agency and the Committee for Medicinal Products for Human Use.48
Statistical analysis
Power calculations could not be performed before the study because of the lack of information from previous studies on the potential effect sizes. In 2005, the prevalence of MS in Cyprus (600 000 population) was 120/100 000. Based on the aforementioned MS patient numbers in our country and the reference centre, the Cyprus Institute of Neurology and Genetics, we were able to enrol 20% of the total RRMS patients eligible for treatment. The sample size was strictly based on the participants’ availability and the novelty of the assessed intervention.
The baseline characteristics were compared across all intervention groups by analysis of variance or the Kruskal-Wallis rank test for continuous variables and by mean Fisher's exact test for categorical variables, as appropriate.
For the primary outcome, the ARR was analysed in a pair-wise fashion for the active interventions compared with the placebo using negative binomial regression models adjusted for the number of relapses within 2 years, the EDSS score at baseline and DMT. The relapse rate was calculated as the total number of relapses divided by the total number of patient-years followed for each treatment group. ARR differences were also calculated among all comparable parameters and reported as the per cent difference.
For the secondary end point, the time to disability progression, the Kaplan-Meier curves were constructed. The progression of disability and time thereof were compared in a pair-wise fashion for the active interventions versus placebo by the log-rank test in the main analysis and by the Cox proportional-hazards models with adjustments for the baseline EDSS score, age and DMT in the supportive analysis. Multivariate models considered all variables with p<0.1 in the univariate models. There was no overt violation of the proportionality assumption.
Both per-protocol and intention-to-treat (ITT) analyses were performed for different sets of research questions to be answered, and both are reported. Missing data of the five patients lost to follow-up were imputed by the last-observation-carried-forward approach. Owing to the proof-of-concept design of the study, the considerable non-adherence rate (49%) and the resulting interpretation issues regarding the ITT analysis, the per-protocol analysis was considered to be the more informative and appropriate method to answer the research questions addressing the efficacy of the interventions when the participants continuously followed the protocol. All statistical analyses were well-defined a priori. All analyses were performed with STATA SE V.10.0 (College Station, Texas, USA). p Values were two-tailed.
Role of the funding source
The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript. All members of the writing group had full access to all study data, contributed to its interpretation and prepared, reviewed and approved the manuscript for submission. All authors had the final responsibility for the decision to submit the paper for publication.
Results
Study population
Among the 80 patients, 20 were randomly assigned to each of the three groups to receive the interventions, and 20 to receive the placebo (figure 2). The baseline characteristics of both the ITT and per-protocol populations were similar across the groups (table 2A and B). All patients who dropped out completed the follow-up until the study completion and were included in the ITT analyses (table 3). Five patients were lost to follow-up before their first scheduled visit. Two other patients who dropped out before their first scheduled visit progressed to secondary progressive MS. Fifteen patients dropped out without successfully completing the ‘normalisation’ period, including five pregnancies. Another 17 patients dropped out early after the entry baseline. Seven patients who dropped out were given monoclonal antibody treatment (natalizumab). Overall, a total of 41 (51%) patients completed the 42-month study, one patient from group A and two from the placebo group transferred to natalizumab, and 39 (49%) patients either withdrew (dropped out) or were lost to follow-up. The reasons for discontinuation are listed in figure 2.
Section A reports the demographics and baseline disease characteristics for the total randomised population by treatment arm and section B reports the demographics and baseline disease characteristics of the all-time on-study population by treatment arm
Section A reports the 2-year primary end point of relapses based on the study design as reported by the dropout patients by treatment arm and section B reports the comparison of the 24-month pretreatment ARR (baseline) with the 24-month on-treatment ARR for the total randomised population by treatment arm

Study flowchart.
Efficacy
Relapses
As a proof-of-concept trial, we primarily needed to answer whether the interventions were effective for those patients who adhered to the assigned treatment, which was the per-protocol analysis.49 For methodological comprehensiveness, we also performed the ITT analysis as a secondary analysis to answer different questions that were complementary to our core hypothesis, such as what happened to all MS patients who were placed on the interventions (the effect of assignment).49
In the per-protocol analysis, during the first year of the treatment, the ARR was 0.80, 0.40, 0.78 and 0.83 for the four intervention groups, respectively. During the second year, the ARR was 0.90, 0.40, 0.67 and 1.25 for the four intervention groups, respectively. Overall, for the 2-year primary end point, eight relapses were recorded for the 10 patients in the PLP10 group (0.40 ARR) versus 25 relapses for the 12 patients on the placebo (1.04 ARR), a 64% adjusted relative rate reduction (RRR) for the PLP10 group (RRR 0.36, 95% CI 0.15 to 0.87, p=0.024; tables 4A and 5 and figure 3A and C). After excluding patients on monoclonal antibody (natalizumab) treatment, the observed adjusted RRR became stronger (72%) over the 2 years (RRR 0.28, 95% CI 0.10 to 0.79, p=0.016, 4B and 5). Pair-wise comparisons for the other two groups against the placebo did not yield significant results (Table 4A and B). The proportion of patients with ≤1 relapse for the 2 years on-study was higher in the PLP10 group than in the placebo group (90% vs 42%, p=0.030, table 5). Seeking to further investigate the observed difference, we compared the relapse rate during the 24 months before the entry into the study to the 24 months on-treatment for each intervention group. We observed a significant relative reduction in the ARR (70%) only in the PLP10 group (RRR 0.30; 95% CI 0.14 to 0.65, p=0.003, table 4A); within-group comparisons for the ARR reduction of the three other groups were not significant and remained not significant when the natalizumab-treated patients were further excluded from the analysis. The effect of PLP10 through time at different time-windows versus placebo for all-time on-study patients is shown in figure 3A–D. Although the ARR analysis within time-windows was not an assigned end point, it could help with the process of evaluating parallel information, such as the efficacy profile through time. PLP10 reached its maximum effect within 1 year on-treatment (counted from the entry baseline) and remained stable afterwards at an ARR of 0.4 with some free-relapse time-windows. Figure 3D demonstrates the dispersion of relapses throughout the 2-year period of all-time on-study (excluding patients on natalizumab) for PLP10 (n=10) versus placebo (n=10). The placebo group, in line with the existing knowledge of how the relapse history works in relation to future relapses in MS patients (contagion phenomenon), showed the expected trend of increased relapse incidences.50 The same phenomenon was true for groups A and C. Finally, during the 12 month poststudy extended period, the on-study patients who received PLP10 showed a persistent benefit in the ARR compared with the placebo (six relapses for the 10 participants within the PLP10 group, 0.6 ARR vs 19 for the 12 participants within the placebo group, 1.58 ARR), indicating a statistically significant 62% adjusted RRR in the ARR for the PLP10 group (RRR 0.38, 95% CI 0.12 to 0.99, p=0.046).
Section A reports the 2-year primary end points of the ARR for the all-time on-study population by treatment arm and per cent difference from the placebo and section B reports the comparison of the 24-month pretreatment ARR with the 24-month on-treatment ARR of the all-time on-study population excluding patients on natalizumab and the comparison of the ARR during the 24-month period on-treatment (primary end point) for each treatment group compared with the placebo
Clinical end points according to study group for the all-time on-study population
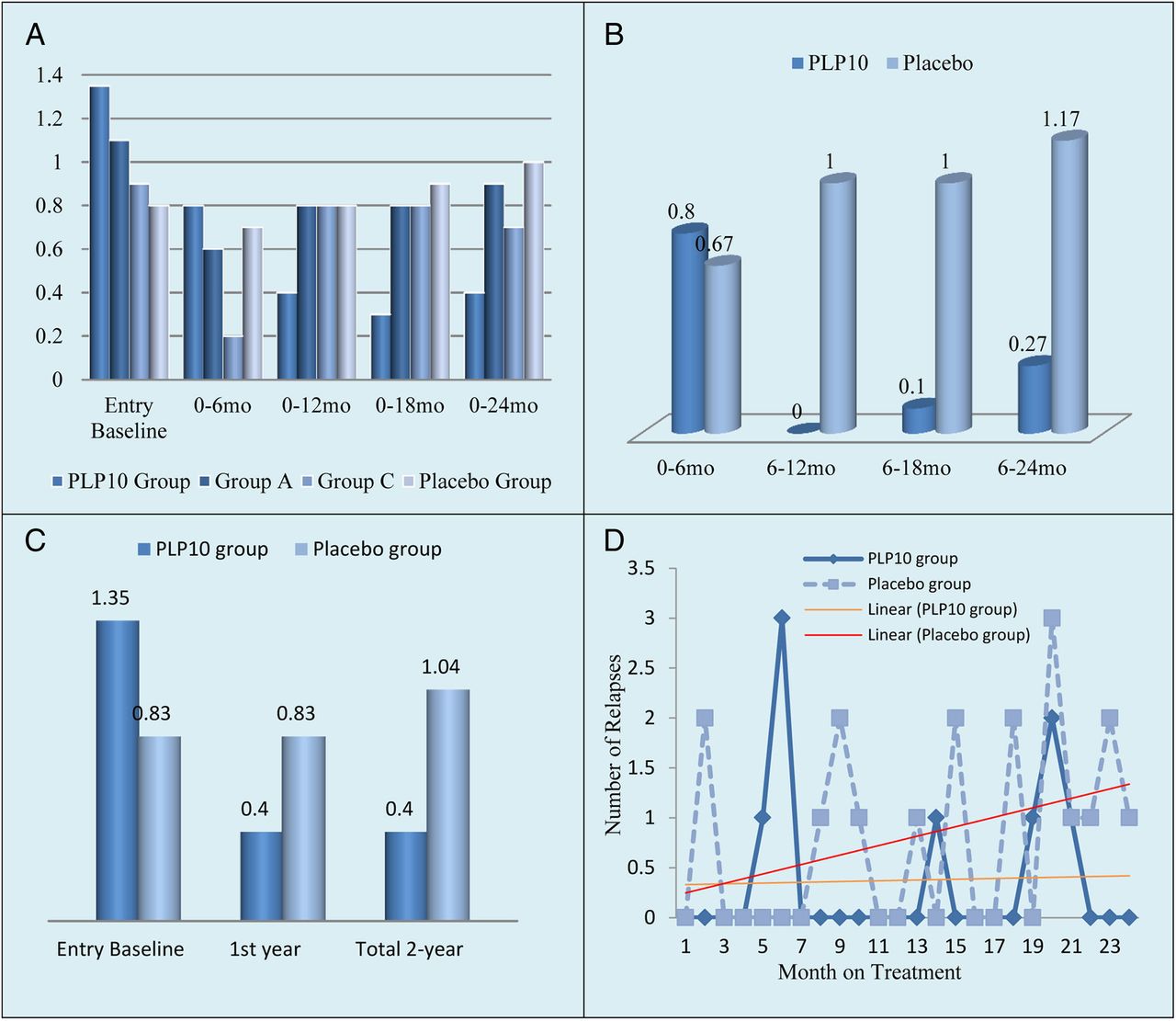
(A) Demonstrates the ARR of the all-time on-study patients during the 24-month pretreatment (baseline ARR) and at different on-study intervals (6, 12, 18 and 24 months) per-treatment arm.* (B) Demonstrates the ARR of the all-time on-study population between the 0–6, 6–12, 6–18 and 6–24-month period intervals for the PLP10 versus placebo groups.* (C) Demonstrates the ARR of the all-time on-study population for the PLP10 versus placebo groups at baseline, during the first year, and during the second year on-treatment.* (D) Demonstrates the dispersion of relapses throughout the 2-year period of all-time on-study (excluding patients on natalizumab) for PLP10 (n=10) versus placebo (n=10). The placebo group showed an irregular dispersion of relapses compared with the PLP10 group, with a linear increasing trend, whereas the PLP10 group showed a stabilised linear trend. Using the per-protocol model in which the patients on natalizumab were excluded, the number of relapses could be compared on the same number of patients.* Including the patients on natalizumab.
Regarding the ITT analysis, the relapses of the dropout patients are reported in table 3A. As expected, no statistically significant differences in the ARR were calculated for the comparison of any group versus placebo for the 24 months on-treatment (table 3B). The ITT population on DMT and/or on natalizumab is shown within the online supplementary information figure 1. Interestingly, despite the high non-adherence rate, there was a statistically significant difference for the comparison of the ARR in the 24 months before entry baseline with the 24 months on-treatment for the PLP10 group (RRR 0.45, 95% CI 0.26 to 0.78, p=0.005).
Disability progression
In the per-protocol analysis, at 2 years, the time to disability progression was significantly longer only with PLP10. The cumulative probability of disability progression was 10% in the PLP10 group and 58% in the placebo group (p=0.019; see online supplementary information figure 2). After excluding the patients on natalizumab, there was again a statistically significant difference between the PLP10 and placebo group for the same analysis (p=0.006; figure 4A). At 2 years, the cumulative probability of disability progression was 10% in the PLP10 group and 70% in the placebo group, which represents a decrease of 60% points or a relative 86% decrease in the risk of the sustained progression of disability within the PLP10 group (adjusted HR, 0.11; 95% CI 0.01 to 0.97, p=0.047). One versus 7 of 10 patients progressed to confirmed disability in the PLP10 and placebo groups, respectively, when patients on natalizumab were excluded. No statistically significant difference was observed for any comparison of the other two groups with the placebo group (figure 4A and see online supplementary information figure 2).

(A) Demonstrates the Kaplan-Meier plot of the time to sustained progression of disability among the all-time on-study patients, excluding the patients on natalizumab, receiving interventions A, PLP10 and C compared with placebo. PLP10 reduced the risk of the sustained progression of disability by 86% over 2 years (p=0.006). Intervention formula A reduced the risk of the sustained progression of disability by 53% (p=0.266), and intervention formula C, by 67% (p=0.061). (B) Demonstrates the Kaplan-Meier plot of the time to sustained progression of disability among the intention-to-treat population receiving interventions A, PLP10 and C compared with placebo. PLP10 reduced the risk of the sustained progression of disability by 71% over 2 years (p=0.052, trend). Intervention formula A reduced the risk of the sustained progression of disability by 22% (p=0.727), and intervention formula C, by 40% (p=0.447).
In the ITT analysis, at 2 years, the cumulative probability of progression was 10% in the PLP10 group and 35% in the placebo group (p=0.052, a trend for an effect), which represents a decrease of 25% points or a relative 71% decrease for the PLP10 group with respect to the risk of sustained progression of disability (adjusted HR 0.22, 95% CI 0.04 to 1.07, p=0.06; figure 4B). Two versus seven of the total randomised patients progressed to confirmed disability in the PLP10 and placebo groups, respectively. No significant differences were observed for groups A or C compared with the placebo group (figure 4B). The mean change in the EDSS score as a function of visit number is shown in figure 5.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean change in the expanded disability status scale score as a function of visit number. The values are expressed as the mean±SE of the mean (s.e.m.), ¶ Including patients on natalizumab and ¶¶ excluding patients on natalizumab.
MRI
Over 2 years, the MRI results supported a PLP10-related positive effect as only 29% from the PLP10 group, in contrast to 67% from the placebo group, developed new or enlarging T2 lesions (57% relative risk reduction). After excluding the patients on natalizumab, there was an increased relative risk reduction (64%) for PLP10 compared with the placebo group, with 29% of patients on PLP10 and 80% on placebo developing new or enlarging T2 lesions (table 5).
Safety
Over the course of the 30-month study, no significant adverse events were reported for any group. The only aetiology for the dropouts was the palatability and smell of the formula preparations in addition to pregnancy. Nausea was reported by two patients. No abnormal values were observed in any of the biochemical and haematological blood tests. No allergic reactions were reported.
Discussion
In this proof-of-concept, randomised, double-blind clinical trial assessing the safety and efficacy of three variations of a novel nutritional formula in RRMS, we observed a significant association for a formula containing a balanced mixture of specific Ω-3 and Ω-6 PUFAs, MUFAs, SFAs, vitamin A, vitamin E and γ-tocopherol (PLP10) compared with the placebo for both the ARR and the progression of disability in the per-protocol analysis. Our results included analyses pertaining to a total of 42 months of study-collected data, including the 12-month intervention-free treatment extension period. We also observed a high dropout rate that was mostly the result of formula palatability, a common phenomenon in trials using oily interventions. Interestingly, a statistically significant reduction in the ARR and disability progression was also observed when comparing the ARR of the PLP10 patients in the 24-month period prior to the study with the ARR of the 24 months on-study; the observed differences became larger when the patients who received natalizumab (currently the most potent disease modifier) were excluded. The ARR decreased within a year on PLP10 and remained stable until the study's completion. The statistically significant difference in the ARR between patients on PLP10 and those on placebo continued for the 12-month extended period (persistent effect) without a significant difference on the DMT. These clinical findings are supported by the results from the MRI analysis, in which the proportion of patients free from new or enlarging brain T2 lesions was also higher in the PLP10 group than the placebo group. No severe side effects have been reported.
To the best of our knowledge, this study is the first randomised clinical trial assessing the proposed combination of active ingredients in a standardised proportion and dosing scheme for MS treatment designed according to the SM approach. Nutrition is commonly accepted as one of the possible environmental factors involved in the pathogenesis of MS, but its role as a complementary MS treatment is unclear and largely disregarded.51 It is well known that the majority of the patients suffering from MS do use dietary supplements for a variable length of time.52 Dietary antioxidants and fatty acids may influence the disease process in MS by reducing immune-mediated inflammation, oxidative stress and excitotoxic damage.12 Published data have revealed that healthy dietary molecules have a pleiotropic role and are able to change cell metabolism and downregulate inflammation by interacting with enzymes, nuclear receptors and transcriptional factors.51 Currently available treatments are the products of reductionism, partially effective and associated with severe side effects. Interferons and glatiramer acetate, the most widely used first-line MS drugs available today, are associated with the least severe side effects among the MS therapies, but they are reported to reduce the ARR only by about one-third and with no significant effect on the progression of disability.53 Natalizumab reduces the ARR by 68% and decreases the possibility of disability progression by 43%, with 57% of patients free of new or enlarging T2 lesions on MRI scans, compared with 15% on placebo.54 Fingolimod is associated with a 54% ARR reduction (without a significant benefit on the progression of disability). Both natalizumab and fingolimod are second-line drugs associated with severe side effects.55
In a review paper in 2009, Mehta reported different clinical studies on interventions formulated based on the individual aforementioned molecular ingredients or on a specific ratio of the aforementioned molecular ingredients for MS treatment, although no one was reported to be using the antioxidant vitamin γ-tocopherol.56 In our study, the choice of the ingredient proportion and dosing scheme was based upon evidence derived from in vivo and in vitro data. In the Western diet, the ratio of Ω-3 to Ω-6 is approximately 1:20–30; in populations that consume fish-based diets, the ratio is approximately 1:1–2.57 ,58 The intervention daily dose was aiming to be, and believed to be, high enough to restore/amplify body-efficient antioxidant activity and ensure cellular membranes lipid profile normalisation (PUFA content) and simultaneously potentiate the involvement of the ingredients in the anti-inflammatory and recovery mechanisms. Dietary fatty acid molecules need an approximately 6-month period to exert their beneficial effect, and this essential parameter was under consideration for the first time in our study design (normalisation period).46 This chronotherapy parameter might be of major importance and is in line with the SM treatment philosophy. We believe that the persistent effect within the poststudy period is in agreement with the very long washout phase reported for Ω-3 fatty acids, especially DHA, to return to the pretreatment values.46 Considering that Ω-3 PUFA supplementation can promote the replacement of AA within the cellular membranes, we can speculate that an increased inflammatory activity can possibly result during the first 6 months of supplementation.
In addition to EPA, DHA, LA and GLA, PLP10 contained limited quantities of other structural/active PUFAs, specific MUFAs (mostly oleic acid) and SFAs (palmitic and stearic acids), specifically to provide a direct source for neuronal cell membrane rehabilitation and for (re)myelination and neuroprotection because these compounds are all major components, precursors and building blocks of any new physiological myelin and cellular membranes in general. Assembly of the correct molecules into the myelin membrane may be especially critical during active synthesis. If these critical constituents are not directly or indirectly available, amyelination, dysmyelination or demyelination may ensue.59 The maintenance of myelin requires continued turnover of its components throughout life.60 ,61
Different factors and molecular entities appear to be part of the possible aetiology for MS, with specific PUFAs and antioxidants found to be key substances related to all known pathogenic and recovery mechanisms. In our study, we further propose that a holistic SM model approach can be applied by synchronised action. First, there is an obvious convenience in administering one formula containing different specific active ingredients. The currently available evidence supports that nutritional interventions would confer a small to medium treatment effect with an accompanying appropriate safety profile.12 ,52 ,56 Combining these specific active ingredients together with γ-tocopherol and other specific active molecules into one stable formulation is expected to enhance adherence while still offering an appropriate safety profile. A similar approach could not be adopted for pharmaceutical interventions with common and severe adverse events, such as those indicated today for patients with MS. Given the advantages of the simultaneous use and that all the included ingredients have proven individually a valid biological plausibility and have been tested in various settings and under various dose schemes, we also assessed the hypothesis that a novel mixture of these ingredients would have a postulated efficacy attained synergistically through different mechanisms of action.52 ,56 Interestingly, the observed magnitude of the treatment effect cannot be explained by adding up the postulated efficacy estimates of the individual ingredients. Findings from in vitro and in vivo studies support this notion of proposed synergy, although this hypothesis can only be taken forward when the observed treatment effect is validated in various settings and in a larger number of patients.
We acknowledge that our study has two considerable limitations: the small sample size and the high dropout rate. Regarding the sample size, one should bear in mind that this study is a small, phase II clinical trial assessing a novel intervention and thus has comparable size in the appropriate literature. Questions taken forward from this trial can be assessed in a larger randomised trial in which appropriate power calculations would be possible, taking into consideration the findings of this study. The adherence of the participants is another limitation of our study, but the total duration of the study that covers a total of 42 months follow-up adds power to the results.48 We acknowledge that we had to deliver the intervention in the way most frequently associated with low compliance, that is, an oral, liquid formula, thus triggering maximum intolerance due to taste. Nevertheless, the observed suboptimal compliance is in accordance with the published literature in which clinical trials assessing liquid fatty acid interventions show a weaker adherence compared with clinical trials of pharmaceutical interventions. Indeed, in our study, we consistently recorded the reasons for withdrawal: most of the participants did not discontinue due to safety issues, but rather due to palatability issues. Controlling non-compliance due to palatability issues is by far easier to address compared with non-compliance related to adverse events and can be resolved when optimisation of the formulation is achieved in future trials. At this stage of the development of the intervention, we would by far exceed the cost-effectiveness threshold if we were to invest in improving these features of the intervention. Moreover, we should also note that MS patients are participant to far more frequent and more serious adverse events related to the current standard treatments.
As a direct consequence of the low compliance and the loss of power, the performed ITT analysis was far less robust than intended, and we would then have to take into serious consideration the performed per-protocol analysis. We focused on the per-protocol data analysis because it is the appropriate method to best provide the answer for the proof-of-concept trial-addressed question.24 To validly incorporate the results of the per-protocol analysis into the interpretation of the overall results of the trial, we need to ensure that the randomisation was not seriously violated due to the exclusion of the non-compliers. The comparison between the baseline characteristics of the patients included in the per-protocol analysis did show a relative balance in the compared groups for known confounders. Nevertheless, the presence of unknown confounders introducing bias to the trial results cannot be excluded despite non-significant differences in the baseline characteristics. As an additional safeguard towards that end, we also performed adjusted analyses for the primary and secondary analyses for important clinical and demographic parameters, that is, relapses, EDSS, age and DMT.
The present preliminary, small-size, randomised, controlled phase II clinical trial provides evidence for a novel nutraceutical formula based on dietary, metabolic, immunological and neurobiological pathways possibly involved with disease progression in MS. This novel intervention showed signs of efficacy in the observed ARR and disability progression. We took the appropriate methodological measures to control for potential sources of bias and to enable a valid interpretation to be reached. We acknowledge that the presence of bias can only be minimised, not excluded, in any clinical research setting and also that random error is always a possible scenario in small trials. Thus, we present the observed results as an additional piece of randomised evidence and anticipate the replication of our study findings in a larger randomised controlled clinical trial.
Acknowledgments
We thank all participating patients. We thank Thyrsos Posporis MD and the central reading centre (Ayios Therissos Medical Diagnostic Centre, Nicosia, Cyprus) as well as Eleni Eracleous, MD (neuroradiologist) for contributing the MRI scans and their teams for the MRI reading. Special thanks to Elena Kkolou, the pharmacist involved in the study, and Eftychia Gaglia for her nursing contribution and collection of blood from the patients. We also thank Demetris Hadjisofoklis and Ioanna Leontiou (University of Nicosia, Helix Business Incubator) for their contributions to the randomisation process, data collection, filing and blind code keeping. Additionally, we would like to thank CING for hosting the project. Moreover, we thank the Cyprus Ministry of Commerce, Industry and Tourism for funding the project and Yasoo Health Ltd. for providing some of the raw materials in exchange of investigating, on their behalf, the efficacy and safety of γ-tocopherol.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
MCP and ISP are joint first and corresponding authors.
-
Contributors All authors interpreted the data. ISP drafted the report and figures, and all authors critically revised and approved the final version. MCP and ISP were responsible for the protocol and study design. MCP was the evaluator of the patients’ clinical progress signs and the treating physician. ISP and GNL were involved in the non-pharmacological treatment-related care. ISP performed the literature search, and both ISP and GNL contributed on the intervention formulation and composition rationale. ISP supervised the composition procedure of the interventions and the fatty acid profile analysis of the red blood cell membranes. EEN and ISP performed the statistical analyses. EEN was an independent scientist. All authors vouch for the accuracy and completeness of the data and the statistical analyses. All authors read and approved the final manuscript.
-
Funding Funding support was by a grant from the Cyprus Ministry of Commerce, Industry and Tourism, as part of a programme for the creation of new high technology and innovation enterprises through the business incubator.
-
Competing interests MCP, GNL and ISP received grant support from the Cyprus Ministry of Commerce, Industry and Tourism, the Program for the Creation of New High Technology and Innovation Enterprises through the Business Incubator. PALUPA Medical Ltd is a research company formed and registered for the completion of the study, as required by the Government’s funding grant programme. MCP, GNL, ISP and CING are all stockholders of PALUPA Medical Ltd. MCP is an employee at the CING. ISP is a senior research collaborator hosted by CING. GNL is a CING collaborator. EEN declares no competing interests. No pharmaceutical companies were involved in this phase II clinical trial. The intervention is under a USA provisional patent; Application Number 61469081.
-
Patient consent Obtained.
-
Ethics approval Cyprus National Bioethics Committee (reference EEΒΚ/ΕΠ/2005/10).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement No additional data are available.