Article Text

Abstract
Objective To explore the structure and content of a non-random sample of clinical study reports (CSRs) to guide clinicians and systematic reviewers.
Search strategy We searched public sources and lodged Freedom of Information requests for previously confidential CSRs primarily written by the industry for regulators.
Selection criteria CSRs reporting sufficient information for extraction (‘adequate’).
Primary outcome measures Presence and length of essential elements of trial design and reporting and compression factor (ratio of page length for CSRs compared to its published counterpart in a scientific journal).
Data extraction Data were extracted on standard forms and crosschecked for accuracy.
Results We assembled a population of 78 CSRs (covering 90 randomised controlled trials; 144 610 pages total) dated 1991–2011 of 14 pharmaceuticals. Report synopses had a median length of 5 pages, efficacy evaluation 13.5 pages, safety evaluation 17 pages, attached tables 337 pages, trial protocol 62 pages, statistical analysis plan 15 pages and individual efficacy and safety listings had a median length of 447 and 109.5 pages, respectively. While 16 (21%) of CSRs contained completed case report forms, these were accessible to us in only one case (765 pages representing 16 individuals). Compression factors ranged between 1 and 8805.
Conclusions Clinical study reports represent a hitherto mostly hidden and untapped source of detailed and exhaustive data on each trial. They should be consulted by independent parties interested in a detailed record of a clinical trial, and should form the basic unit for evidence synthesis as their use is likely to minimise the problem of reporting bias. We cannot say whether our sample is representative and whether our conclusions are generalisable to an undefined and undefinable population of CSRs.
- Medical Ethics
- Medical Journalism
- Internal Medicine
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Article summary
Article focus
-
What are clinical study reports (CSRs)? What do they contain and how long are they?
-
Can CSRs help address reporting biases associated with the published literature, and improve the quality of evidence synthesis?
Key messages
-
CSRs represent a hitherto hidden and untapped source of detailed randomised controlled trial data (mean page length: 1854 pages), increasingly becoming publicly available, and should form the basic unit for evidence synthesis to minimise the problem of reporting bias.
-
CSRs show that numerous individuals make important technical contributions to the design, conduct and reporting of each trial, but journal publications often fail to record these details, resulting in a loss in individual responsibility for what is reported.
-
The ICH E3 guideline to which most CSRs conform was published in 1995, and needs updating.
Strengths and limitations of this study
-
We cannot say whether our sample is representative and whether our conclusions are generalisable to an undefined and undefinable population of CSRs.
Introduction
Systematic reviews are thought to provide one of the most robust ways to evaluate the effects of healthcare interventions. But the robustness of findings clearly rests upon reviewers having sufficient access to clinical trial information to critically evaluate and reproduce the original research. Research on reporting bias over the last decades has shown that trusting the published literature at face value, even peer-reviewed publications, can be fraught with difficulty—a problem that spans drug classes.1–12
Following the decision by the European regulator, the European Medicines Agency (EMA) on 30 November 2010, to make available a broad spectrum of documents related to medicinal products for human and veterinary use,13 ,14 attention has focused on one particular type of regulatory document—clinical study reports (CSRs).15–18 CSRs are usually written for regulators following guidelines developed by the industry regulatory collaborative effort ‘International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use’ (ICH). The ICH guidelines ‘structure and content of clinical study reports’19 (see online supplementary appendix 1) are known by the document code ‘E3’. They were formalised in 1995 ‘to assist sponsors in the development of a report that is complete, free from ambiguity, well organised and easy [for regulators] to review’.19 E3 has not been edited or changed since 1995.
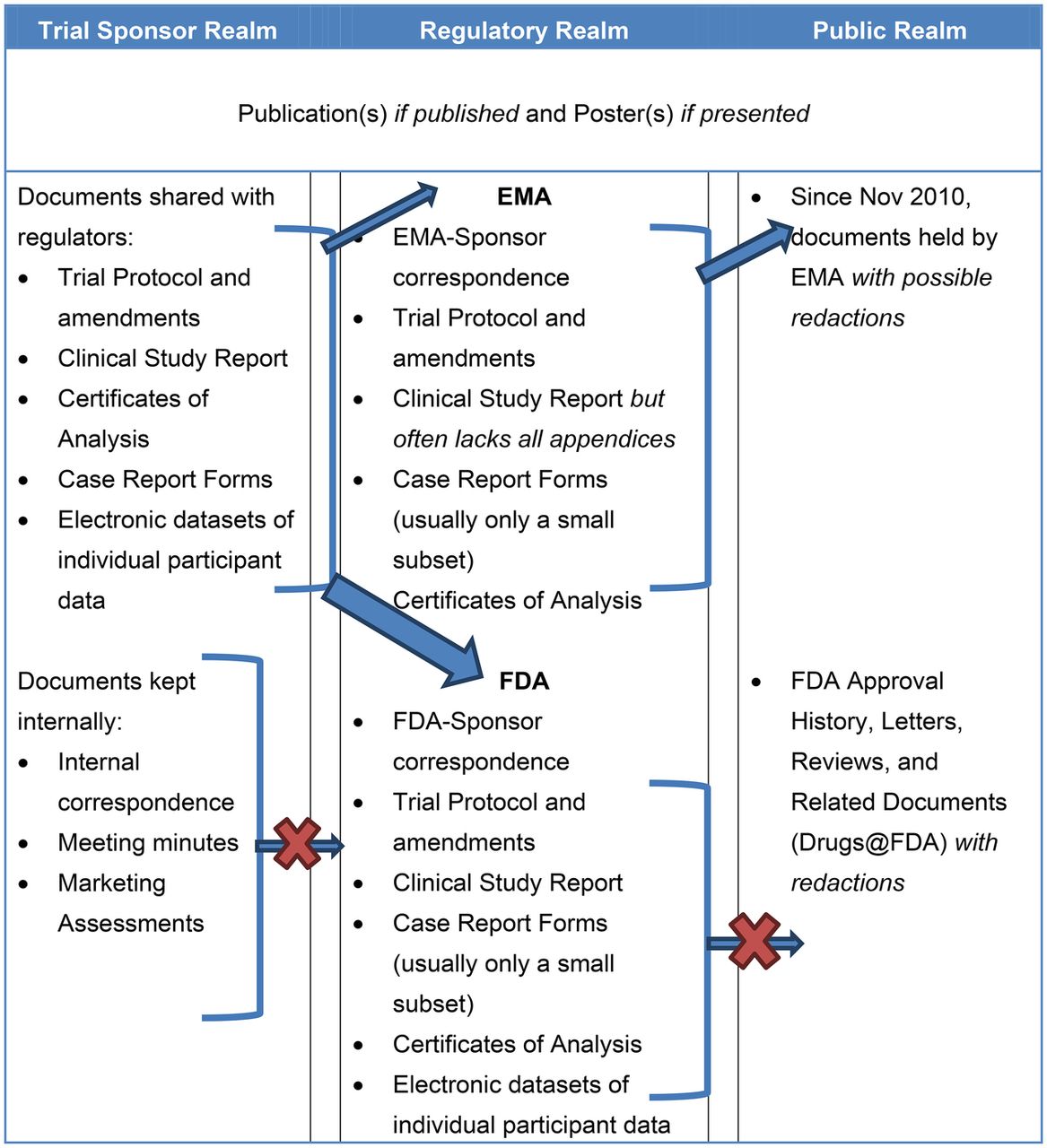
CSRs are but one category of information that is transmitted from study sponsors to regulators (figure 1), but are important as they contain substantially more information and detail on the intervention being tested than published versions of the same trial. The wealth of information may be sought with increasing frequency by researchers appraising single trials, entire trial programmes, or by those synthesising evidence.17 ,20 We are aware of two recent examples of systematic reviews of the effects of pharmaceuticals carried out using CSRs and other regulatory material.12 ,21 One group also concluded that journal publications insufficiently report clinical trials.22
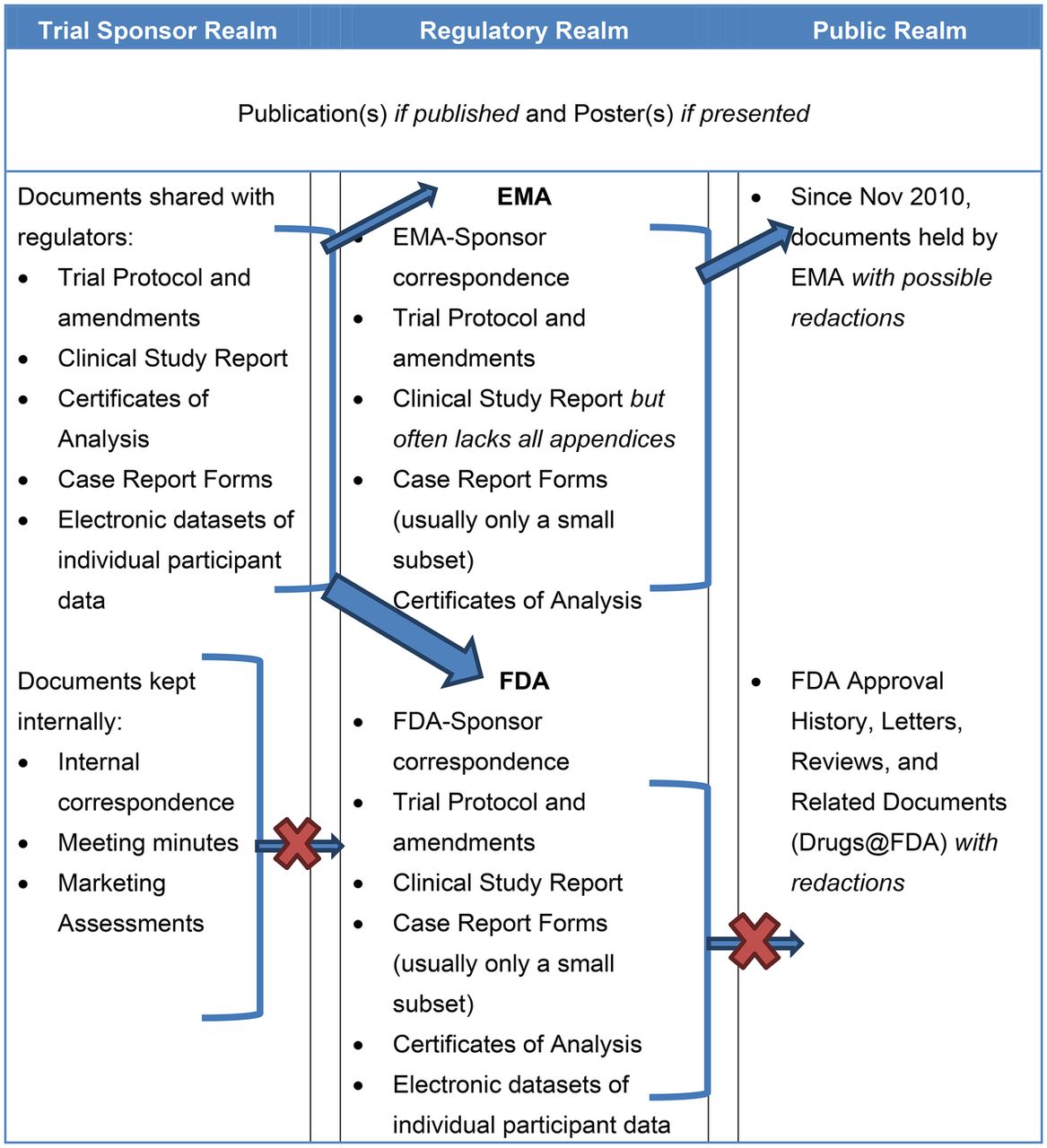
Types of clinical trial data typically held within and transferred between three realms: trial sponsor, regulatory and public.
Despite CSRs’ potential importance little is known about their structure and content outside of those individuals with direct involvement in regulatory processes. This knowledge gap may hinder development of methods for fair and reliable appraisal of CSRs and their use in evidence synthesis. We are not aware of any instruments specifically designed for appraising CSRs. Lack of visibility may also hinder understanding of the complexity of the organisation and the reporting of clinical trials.
We carried out an exploratory review to describe the structure and content of a non-random sample of CSRs. By describing the contents of CSRs, this research seeks to transform CSRs from an obscure document only known to regulators and industry into a more widely known and accessible document. Our long-term intention is to improve the credibility of research synthesis by facilitating a move from the level of detail found in journal articles to the level of detail found in regulatory documents, thus guiding clinicians and other decision makers at all levels.
Methods
We obtained CSRs from public sources, as follows:
-
Requesting from EMA, under its Freedom of Information (FOI) policy, CSRs for manufacturer-sponsored trials of the 10 best-selling prescription-bound products in the USA in 2010.23
-
Reusing CSRs from our own previous research (oseltamivir and zanamivir).12
-
Downloading CSRs openly available on the Internet. Search terms were not predefined, but sites searched included Google (http://www.google.com), the Drug Industry Document Archive (http://dida.library.ucsf.edu/) and Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (Institute for Quality and Efficiency in Healthcare) (IQWiG)'s library of reboxetine studies (https://www.iqwig.de/information-on-studies-of-reboxetine.980.en.html).
-
Corresponding with one researcher who obtained CSRs through an FOI request to Food and Drug Administration (FDA) (epoetin alfa).
-
Requesting manufacturers fill any gaps in the completeness of reports that we believe are legally required to be publicly available (paroxetine).
To create as broad a database as possible, we did not apply restrictions in drug type or family or sponsor. We did not submit requests under the Freedom of Information Act to the FDA, because such requests can take years to be fulfilled and—if fulfilled—may be heavily redacted.24
We did not draw a random sample of CSRs as there is no known sampling frame. No one knows how many reports have been written by intervention category as there is no central register of CSRs. Through familiarity with CSRs for oseltamivir and zanamivir, which were included in one of our Cochrane reviews,12 we developed and piloted a data extraction sheet designed to capture the salient characteristics of CSRs. We created a list of around 40 potential sections we expected to find, generated directly from elements specified in E3. For each element in the list, we checked whether the obtained CSRs included that section (confirmed either by direct identification of the section or an indication the section existed based on the CSRs’ table of contents), whether we had access to it and its page length. Because of previous difficulties we had accessing CSRs appendices, we also recorded whether sections were listed as appendices or not. The page length was calculated either by directly counting the pages or by estimating their size from the table of contents of each report, and was used as a crude proxy for the level of detail available. The page lengths were rounded up to the next integer, and were summarised by reporting medians and ranges. We also included questions relating to trial registration and authorship. Our (blank) data extraction sheet is in online supplementary appendix 2.
All variables from CSRs were first extracted in single. We subsequently audited each other's extractions, checking the accurateness of the information. We chose to present elements analogous with those that typically appear in trials reported in scientific journals including the study synopsis (a brief summary of the study), the study protocol (written prospectively, describing the study methods), efficacy and safety evaluations (a narrative summary of the efficacy and safety results of the study, including tables and figures), as well as attached tables. We also included elements rarely found in journal publications: sample (blank) and completed case report forms (CRFs are paper or electronic forms designed to capture prespecified efficacy and safety related information for each study participant), the statistical analysis plan (a prospectively written narrative and/or statistical code indicating how trial data will be analysed) and individual participant efficacy and safety listings. The corresponding E3 section numbers are listed in table 2. Disagreements were resolved by discussion.
Our uncorrected (original) and corrected extraction sheets as well as audit records are available upon request from the corresponding author.
We calculated a compression factor for published trials which we defined as the ratio of CSR page length compared to the page length of the same trial as published in scientific journals. The objective of this metric was to convey a rough sense of how much information present in CSRs may be condensed (‘compressed’) in short journal publications, in consideration of CSRs’ far greater length and level of detail. The size (page length) reflects the level of detail as well as the presence of many elements such as protocols and their amendments, randomisation lists, statistical analysis plans, certificates of analysis and extra data on subpopulations. We have demonstrated12 that these elements are essential for understanding and appraising a trial. The compression factor is a crude measure of how much is compressed or simply left out of each publication which will affect the reliability of the appraisal and interpretation of trials. Trial publications were searched for in multiple sources—clinical trial registers, published systematic reviews and correspondence with sponsors. Because in most cases we could not access all parts of all CSRs (and therefore do not know their complete page length), we calculated ‘conservative’ compression factors as well as ‘realistic’ compression factors. ‘Conservative’ compression factors were calculated on a trial by trial basis using the total number of pages in CSRs available to us divided by the length of journal reports for that same trial, whereas ‘realistic’ compression factors were based on the true total page length of the CSRs.
Results
We identified 84 documents believed to be CSRs for 14 compounds. These covered therapeutic and biological interventions including antipsychotics, antidepressants, antivirals, natural antiarthritics, anti-inflammatory agents, pandemic influenza vaccines, statins, erythropoietins and antiplatelet compounds. We included English-language summaries of two Japanese oseltamivir studies (JV15823 and JV15824) as they had been presented to EMA in this form. We excluded documents which were sections of CSRs that nonetheless contained insufficient information to understand the overall content of the CSRs (olanzapine F1D-LC-HGAV, F1D-MC-HGAJ and F1D-MC-HGAO) and three documents which we had originally classified as CSRs but were not (reboxetine 14, 22 and 37). This left us with 78 CSRs (144 610 pages) (figure 2). The median pages obtained per CSR was 644 (range 9–15 440). Only 4 of 78 CSRs (reboxetine 8, 16, 17 and 91) were written prior to 30 November 1995 when ICH E3 was approved. Table 1 summarises the pharmaceutical, manufacturer, date and provenance of the CSRs in our review. EMA reported not holding studies for esomeprazole magnesium (Nexium), Advair Diskus, quetiapine fumarate (Seroquel), montelukast sodium (Singulair), epoetin alfa (Epogen) and simvastatin.
Pharmaceutical, trials, producers, dates and sources of CSRs in the review

{kind=link}
{kind=link}
Study flow.
All of the 78 included CSRs contained a synopsis (median page length 5 pages). The efficacy evaluation was identifiable and directly accessible in 76 (97%; median length 13.5 pages) and safety in 77 (99%; median length 17 pages). The attached tables were likewise present in 63 (81%) CSRs, with a median of 337 pages long (range 1–3665). Seventy-three CSRs (94%) reported including the study protocol. In the 40 protocols we could access, the median page length was 62. We found blank CRFs included in 68 (87%) CSRs. Of the 33 blank CRFs we could directly access, the median length was 133 pages (range 14–981). For completed CRFs, 16 (21%) reports made direct mention of a section on completed CRFs, but we had access to completed CRFs in only 1 case (Arthronat; length 765 pages).
Fifty-five (71%) of 78 included CSRs included a statistical analysis plan in some form. Of those for which we could directly access the content (n=37), the median page length was 15 (range 3–85). Individual efficacy and safety listings were included in 53 (69%) and 62 (81%) CSRs, respectively. The median page length was 447 (range 15–21 698) for efficacy and 109.5 (range 2–10 954) for safety.
A summary is presented in table 2.
Key characteristics of the clinical study reports in the review
All trial reports in our review were sponsored by the pharmaceutical industry.
Median conservative compression factors ranged between 1 and 1221. The realistic compression factors calculated for the Arthronat, paroxetine and Clopidogrel versus Aspirin In Patients at Risk of Ischaemic Events (CAPRIE) trials were 379, 1021 and 8805, respectively (table 3).
Conservative and realistic compression factors
Discussion
We collected and described a sizeable number of CSRs written in the last two decades. All CSRs contained a table of contents (as specified in E3 section 3); this, together with optical character recognition (to enable searching the full text of the scanned documents) and the occasional need to combine multiple files to create a single document, substantially improved the ease of navigating CSRs.
Despite the size of our non-random sample, it is unclear whether our conclusions are generalisable to all other CSRs. This is because we have extremely limited knowledge about the total population of CSRs in regulators’ and sponsors’ possession. Nevertheless, within our sample spanning different manufacturers, therapeutic classes and times, we found that the structure of CSRs was, within different house styles of presentation, strikingly similar, probably owing to the guidance by ICH E3.25 This suggests that the structure and content of other CSRs is likely to be similar.
The future basic currency of research synthesis?
The median length of 644 pages for reports in this study, as well as CSRs’ routine inclusion of trials’ protocol, statistical analysis plans and blank case report forms, strongly suggests that CSRs are the most detailed and complete, integrated form of reporting of the design, conduct and results of clinical trials. In a study that directly compared the adequacy of reporting between journal articles and CSRs, the authors found that complete information regarding greater than 40% of methods items were only available in CSRs.22 The level of detail found in CSRs thus far surpass the level of detail available in journal publications, and as such they are prime candidates for the next basic currency of evidence synthesis and appraisal of a trial. Given the EMA's new policy of making such documents publicly available, access to these documents is now relatively straightforward.26 However, including CSRs in systematic reviews is labor-intensive, given their size and complexity.12
Accessing complete CSRs
Although CSRs may trump other forms of trial reporting in the public domain (such as conference abstracts or journal publications), serious limitations remain. Despite obtaining 144 610 pages for 78 CSRs, in almost all instances, we lacked complete access to the CSRs’ numerous appendices. Even for the sole complete CSR we obtained (Arthronat MA-CT-10–002), case report forms were provided for only 20% of participants. The Arthronat text does not provide a reason for this omission, but it reflects the vagueness of the relevant section of the E3 guidance (16.3.2) which does not define ‘other CRFs submitted’. Also, we could only access the original trial protocol in 40 (51%) of 78 CSRs obtained. This is important because trial protocols, written prior to patient enrollment in a trial, are an important way to guard against reporting biases.27 ,28
We could obtain individual patient listings in only a minority of cases despite confirming their inclusion in the majority of CSRs (table 2). This may be a significant limitation, as the E3 specifies that ‘the report with its appendices should also provide enough individual patient data, including the demographic and baseline data, and details of analytical methods, to allow replication of the critical analyses.’19 Unavailability was possibly owing to the fact that EMA allows manufacturers to submit CSRs omitting a number of appendices including individual patient data and case report forms (which EMA states should be available within 48 h, if requested).29 In the case of oseltamivir, the primary drug analysed in a Cochrane review we conducted,12 the manufacturer refused to share with us report appendices not submitted to EMA,30 and EMA declined requesting them on our behalf.8 Although FDA likely possesses more complete CSRs and patient-level data, it historically has treated such data as trade secret and/or confidential.31–33 EMA is therefore at present the only reliable source of obtaining CSRs. As such, despite European regulators’ progressive stance—announcing that ‘clinical trial data should not be considered commercial confidential information’34—the completeness gap is unlikely to be filled any time soon.
Another significant limitation is that CSRs are only written for therapeutic, prophylactic or diagnostic agents, and therefore inadequacies remain in evidence synthesis of other types of interventions such as surgical or behavioural interventions.
Individual participant listings
Individual participant listings—which identify participants by a unique ID—were accessible in 29 of the 78 CSRs we reviewed. But these data are difficult to analyse because they are presented as database printouts rather than in the original computer data files. This is understandable considering that CSRs are a written/archival format, but because EMA does not accept SAS format data files,35 ,36 the industry standard, third-party access to databases of patient-level data remains elusive. We see no compelling reason why all regulators should not request these from sponsors and make them publicly available. Whether availability of individual listings and CRFs, with its attendant laborious analysis, would increase our understanding of the trial and its results is unclear. But there is at least one case where the reanalysis of CRFs added invaluable knowledge to that already available in CSRs.37
The public–private debate
One manufacturer has claimed that the non-release of case report forms is motivated by concerns over protecting participants’ confidentiality.38 Nothing we have seen so far corroborates this claim, however an ongoing EMA working group is specifically discussing issues related to protecting participants’ confidentiality. Based on current document releases and position statements, however, it appears that EMA has deemed case report forms and individual patient listings to be, in principle, releasable in their entirety (after a preliminary review).39 Furthermore, individual patient listings are intended to duplicate information contained in filled case report forms. The release of case report forms would ensure the accuracy of individual patient listings with little additional risk to patient confidentiality. Moreover, extra checks such as registration of protocols by bona fide research groups could deter any inappropriate use. We also believe that the sheer bulk of the forms act as a deterrent against malice.
Size matters
Our range of compression factors shows the scale of selection and synthesis which must (consciously or unconsciously) occur in the process of transforming CSRs into journal-length articles. We found a strong resemblance in detail, page length, structure and purpose between the short synopsis section of CSRs and reports of trials as published in scientific journals. In some cases essential items of information such as the trial protocol and its subsequent amendments are simply not included in journal articles or are replaced by methods written post facto. In other cases of items essential for the interpretations of the trial results (such as the statistical analysis plan), tens of pages are reduced to a paragraph on sample size calculation in the journal report, underscoring the lack of detail (and its attendant problems) common to public forms of trial reporting. For example, the ratio of words in the protocol of the CSR for aripiprazole CN138135 to the methods section for published journal article of the same trial is 30.5 (53 713 words in the CSR protocol vs 1763 words in the journal article). For the oseltamivir WP16263 trial, the ratio was 22.7 (26 761 words in the CSR protocol and amendments vs 1177 words in the journal article).
This compression of information also occurs in databases not restricted by length, such as ClinicalTrials.gov.40
Our study raises the question of why the medical community has accepted the low (summary, aggregate) level of detail found in most peer-reviewed journal publications compared with the depth of detail available in CSRs. European regulators recently noted: ‘documents that provide critical information on a study, such as the protocol (16.1.1), statistical methods (16.1.9), list of investigators and study sites and sample case report forms, would always be needed by reviewers assessing a study.’41 Why have those outside of the regulatory world tolerated journal publications lacking such details?
One possibility may be that while the clinical trial enterprise has changed dramatically in the last half century, the scientific journal publication model has not. Since the 1950s, there have been considerable transformations in the political economy of clinical trials driven by the increasingly commercialised and global nature of the pharmaceutical industry, the rise in academic-industry ‘partnerships’ in medicine and increased communication among regulators. It is now common to find trials with study centres scattered around the globe. This increasing complexity and the need to provide an audit record is reflected in the comprehensive tomes documenting the trials—CSRs—but trial reporting in scientific journals remains limited to summary and aggregate details. It should be noted, however, that many journals now have websites which enable them to make available extended content beyond what traditionally appears in the printed journal.
Authorship or contributorship?
Examination of CSRs revealed scores of important technical contributions to the design, conduct and reporting of each trial. These included contributions from database programmers, records officers and CSR writers, often invisible in the published journal article. In some cases, we found no mention in CSRs of individuals who figured as authors of subsequent published trial reports while individuals named as CSR authors went unacknowledged in journal publications. Current International Committee of Medical Journal Editors (ICMJE) guidelines on authorship and contributorship are largely focused on ensuring those placed on by-lines deserve to be authors. But the guidelines also suggest that ‘all contributors who do not meet the criteria for authorship should be listed in an acknowledgements section’.42 Given the complexity of clinical trials, the ICMJE should call for itemised contributorship: the names of all contributors to be specified along with their role in the design, conduct, analysis or reporting of the trial. If the contribution to the trial of most people goes unrecorded, so does their individual responsibility for what is produced. Itemised contributorship records, to all phases of a trial, could be piloted in trial registers.
E3 guidance
The E3 guideline set an excellent standard, but it needs formal updating and further development. For example, there should be a self-standing set of definitions for terms such as ‘CRFs’ and ‘other CRFs submitted’ (section 16.3.2) and a description of how a particular trial fits within a sponsor's trial programme of pharmaceutical development. Apparently forgotten items such as certificates of analysis (describing the appearance and content of the interventions being tested) and post-1995 details such as trial registration numbers should be mentioned.
We hope our review has given CSRs what they have lacked so far: visibility. CSRs represent a largely untapped source of detailed data that we believe can serve as a means of addressing the ravages of reporting bias in all its forms, leading to a more accurate understanding of the effects of medicines.
Acknowledgments
We thank Dr Vallance and Dr Kraus of GlaxoSmithKline for making public selected report appendices from the nine paroxetine trials. We also thank Daniel Coyne for sharing the CSR that FDA sent him in response to his Freedom of Information request, and Iain Chalmers for guidance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online appendix 1
- Data supplement 2 - Online appendix 2
Footnotes
-
Contributors PD and TJ both made substantial contributions to the conception and design of this study. Both authors acquired data analysed in this study, and were involved in the interpretation of the data. PD and TJ drafted and revised the article together, and both approve the final version.
-
Funding Publication of this article was funded in part by the Open Access Promotion Fund of the Johns Hopkins University Libraries.
-
Competing interests All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that: both authors are corecipients of a UK National Institute for Health Research grant to carry out a Cochrane review of neuraminidase inhibitors (http://www.hta.ac.uk/2352). TJ was an ad hoc consultant for F. Hoffman-La Roche Ltd in 1998–1999. He receives royalties from his books published by Blackwells and Il Pensiero Scientifico Editore, none of which are on clinical study reports. He is occasionally interviewed by market research companies for anonymous interviews about Phase 1 or 2 products unrelated to products in this review. From 2011 to 2012 he has acted as an expert witness in a litigation case related to one of the compounds in the review (oseltamivir). He is on a legal retainer for expert advice on litigation for influenza vaccines in healthcare workers. PD received €1500 from the European Respiratory Society in support of his travel to the society's September 2012 annual congress where he gave an invited talk on oseltamivir. Peter Doshi is funded by an institutional training grant from the Agency for Healthcare Research and Quality #T32HS019488. AHRQ had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Both authors’ spouses and children have no financial relationships that may be relevant to the submitted work.
-
Provenance and peer review Not commissioned; internally peer reviewed.
-
Data sharing statement The original extraction forms and audit record are available on request from the corresponding author.