Article Text

Abstract
Introduction There are unmet mental health needs of depressed adolescents and young adults (AYAs) across the USA. Behavioural technology adequately integrated into clinical care delivery has potential to improve care access and efficiency. This multisite randomised controlled trial evaluates how a coach-enhanced digital cognitive behavioural intervention (dCBI) enhances usual care for depressed AYAs in paediatric practices with minority enriched samples.
Methods and analysis Participants (n=750) ages 16–22 who meet threshold criteria for depressive severity (Patient Health Questionnaire-9; PHQ-9 score 10-24) will be recruited through paediatric practices across three academic institutions (Boston, Pittsburgh and San Diego). Participants will be randomised to 12 weeks of dCBI+treatment as usual (TAU) (n=450) or TAU alone (n=300) in outpatient paediatric practices. Assessments will be completed at baseline, 6 weeks and 12 weeks with the primary outcome being improvement in clinician-rated and self-reported depressive severity (Children’s Depression Rating Scale—Revised and PHQ-9) and secondary outcomes being self-reported suicidal ideation (item 9 on PHQ-9), anxiety severity (Generalised Anxiety Disorder), general quality of life (Satisfaction with Life Scale) and general functioning (Children’s Global Assessment Scale). The study design is an intent-to-treat mixed effects regression with group, and covariates nested within the sites.
Ethics and dissemination All participants or their parent/guardian (under 18 years or unemancipated) will give informed consent to a study team member. All data are expected to be collected over 18 months. The Institutional Review Board (IRB) is a board at each institution in the United States that reviews and monitors research involving human subjects. IRB approval from the University of Pittsburgh was obtained on 30 November 2021 (STUDY21080150), from the University of California San Diego’s Human Research Protection Program IRB on 14 July 2022 (802047), and from the Boston Children’s Hospital IRB on 25 October 2022 (P00040987). Full study results are planned to be published within 2 years of initial study recruitment (October 2024). Dissemination of findings will occur in peer-reviewed journals, professional conferences and through reports to participating entities and stakeholders.
Trial registration number NCT05159713; ClinicalTrials.gov
- telemedicine
- mental health
- depression & mood disorders
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
The study design allows for the evaluation of the clinical value-add of a digital behavioural intervention for depression nested in outpatient paediatric care.
The primary outcome is a blinded clinician-rated change in depressive severity, reducing bias.
Participants include a racially diverse sample of adolescents and young adults.
Potential heterogeneity may occur in treatment as usual.
Participant attrition at follow-up is possible, which may contribute to selection bias associated with differences attributed to the two treatment arms.
Introduction
Mental health disorders are associated with increased morbidity, mortality and healthcare costs.1 2 Behavioural health issues in adolescents and young adults (AYAs) are common and have worsened during the COVID-19 pandemic.3 The recent surge in morbidity has strained traditional resources leading to delayed access to care.4 Early intervention utilising tiered modalities to complement traditional treatment is crucial to improving the health and future of our youth.5
Many evidence-based behavioural health treatments exist, with cognitive behavioural therapy (CBT) as one that is effective in treating anxiety and depression.6 When treating AYAs with mild to moderate anxiety and depression, foundational CBT skills, such as cognitive reframing, behavioural activation and problem-solving improve symptoms in this age group.6–9
Despite the evidence, there are often few CBT resources to help treat AYAs with anxiety or depression. Furthermore, many AYAs do not follow through with treatment referrals or adhere to behavioural recommendations.10–12
Given the challenges in access to empirically supported behavioural therapies, many AYAs seek new modalities, including digital cognitive behavioural interventions (dCBI) to address access and use barriers. Studies have shown that dCBIs are as effective as standard treatment in improving anxiety and depression; particularly when coach facilitated.13 14 However, many dCBIs are only available commercially and without medical oversight or in research settings.
Objectives
This study has been designed as a multisite randomised controlled trial (RCT) comparing digital CBT+treatment as usual (TAU) to TAU alone in outpatient paediatric practices. The study will target patients ages 16–22 who meet threshold criteria for depression by scoring 10–24 on the Patient Health Questionnaire-9 (PHQ-9), a validated tool used to identify patients at risk for depression.15 The primary outcomes will be blinded clinician-rated Children’s Depression Rating Scale—Revised (CDRS-R) and the self-report PHQ-9, and secondary outcomes will include self-reported suicidal ideation ((SI) item 9) on the self-reported Patient Health Questionnaire (PHQ-9), Generalised Anxiety Disorder (GAD-7) and Satisfaction with Life Scale (SWLS) at 12 weeks post-treatment among patients randomised to TAU compared to those randomised to TAU+dCBI. The study is specifically interested in changes in self-reported depressive severity, passive SI, anxiety and quality of life and functioning. Moderators of treatment response will be explored, such as adherence to treatment, severity of behavioural health symptoms, race, ethnicity, socioeconomic class and geographical location of the participant.
Aims
Aim 1 is to test the efficacy of digital CBT to reduce both self-report (PHQ-9) and clinical-rated (CDRS-R) symptoms of depression at 12 weeks (primary time point) (6 weeks assessment as secondary time point). An exploratory subaim will examine the correlations between the two measures (PHQ-9 and CDRS-R) at each time point and the extent to which changes in the PHQ-9 are associated with changes in the CDRS-R.
Aim 2 is to examine if there is a reduction in passive SI (PHQ-9 #9), anxiety symptoms (GAD-7) and improvement in quality of life and functioning (SWLS and Children’s Global Assessment Scale (CGAS)).
Aim 3 is exploratory and is to identify baseline characteristics that moderate treatment response and/or predict treatment adherence. Key baseline characteristics include race, socioeconomic class, geography (site) and participant expectancy for improvement (Credibility and Expectancy Questionnaire). The baseline characteristics were selected due to the expected diverse population across the three sites.
Methods and analysis
Scientific Advisory Board
Scientific Advisory Board committee members will include mental health providers, researchers, subject matter experts and paediatricians. The board will monitor study progress relative to the goals and milestones, review study deliverables and provide strategic guidance and direction of the research.
Setting
The investigation will occur in three geographically diverse paediatric primary care practices: Children’s Hospital of Pittsburgh, Boston Children’s Hospital and Rady Children’s Hospital of San Diego, with Pittsburgh serving as the coordinating centre (see figure 1 for proposed study flow). The participating paediatric clinics are representative of the targeted patient population. Prior to selecting study sites, each institution completed a questionnaire with details about their patient populations, clinic structure, triage and therapy protocols and available resources. The participating sites were selected based on site characteristics and patient populations.

Study flowchart. dCBI, digital cognitive behavioural intervention; GAD7, Generalised Anxiety Disorder-7; PHQ9, Patient Health Questionnaire-9.
Study population
Eligible participants are AYAs (age 16–22 years) who have at least moderate depressive severity and are patients at one of the three participating sites. Using a permuted block randomisation scheme, the study plans to randomise 250 participants per site (150 randomised to dCBI with TAU and 100 to TAU alone) for a total of 750 participants.
Patient recruitment and eligibility criteria
Paediatric clinics at all three sites are the source for recruitment based on a previous convenience sample noting similar patient characteristics as targeted and previous research performance. Minority sample enrichment will occur.
Patients who score 10-24 on the PHQ-9 (which suggests at least moderate depression) will be introduced to the study by their paediatric clinician or integrated health therapist (IHT). If the patient is under 18, the clinician will introduce the study to both the patient and parent/guardian. If the patient and parent or guardian (if appropriate) is interested in the study, a member of the research team will contact the patient to explain the study in more detail and complete further screening measures after consent (see online supplemental file for informed consent document). This study will only include English-speaking AYAs with access to a smartphone. Patients will be excluded from the study if they have had a current suicide attempt in the past 3 months or current severe psychiatric disorder of bipolar disorder, current substance misuse or dependence, thought disorder, or patients who score greater than 24 on the PHQ-9.
Supplemental material
TAU control group
Participants randomly assigned to the TAU group will receive standard care, consisting of a tiered stepped care model of behavioural therapy if deemed necessary. This is by the IHT or clinical team at each practice, or referral to outside therapists as part of routine care, with the provision of augmentation of therapy (or addition of an antidepressant) at the discretion of the clinical team. Therapists at each site receive standardised education and supervision with quality assurance measures in place as part of routine clinical care. Psychotropic medications and previous behavioural treatment will be recorded at baseline. Number of therapy sessions, and addition of antidepressant or other psychotropic medication and/or dose changes over the study period will also be measured.
Intervention group
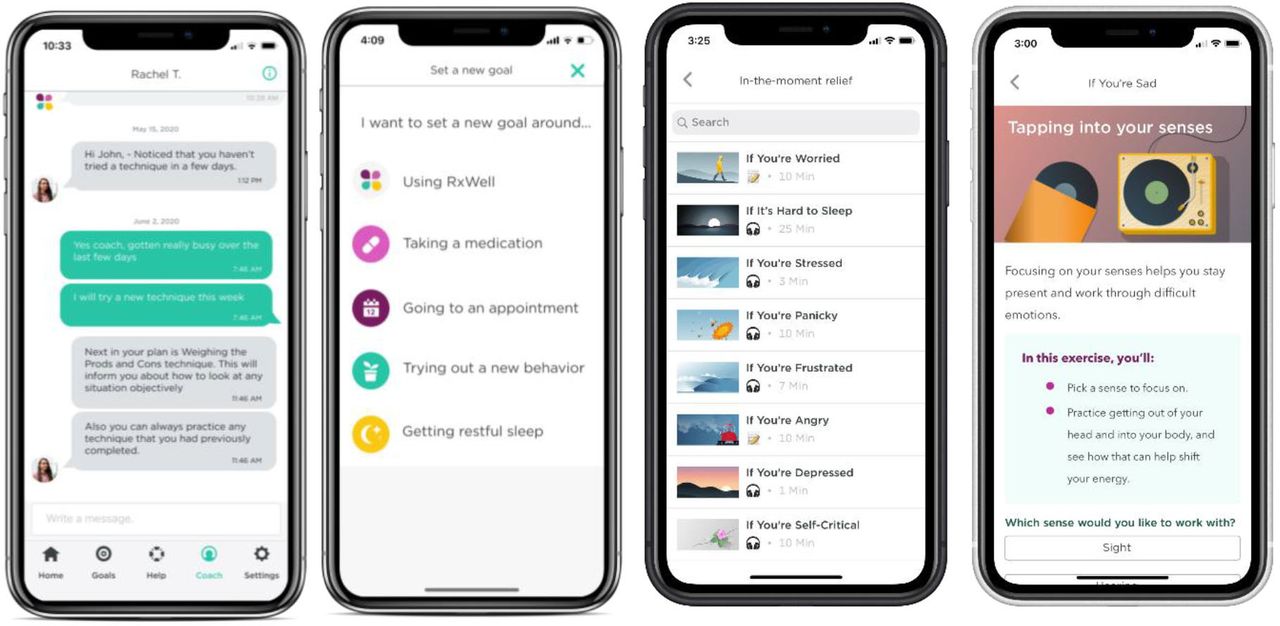
Participants randomly assigned to the intervention group (dCBI+TAU) will receive TAU and additionally will have access to the dCBI. The dCBI uses a platform called RxWell, a trans-CBT mobile app product addressing depression and anxiety that was developed based on standard CBT techniques. RxWell combines health coaching support with evidence-based techniques. RxWell provides users with brief (5–10 min) skill building techniques such as relaxation, behavioural activation and exposure, distress tolerance, cognitive reframing and mindfulness meditation, for anxiety and depression. Users have access to a goal-setting tab and ‘in the moment relief’ section, which contains over 17 techniques, 14 of which are brief audios to help users engage relaxation responses (see figure 2). The user will use the depression path, and if they express significant anxiety; the coach can personalise the programme by pulling in any of the 53 techniques from the anxiety path. Clinicians will participate in a training about how to introduce the dCBI and explain to participants that the dCBI is most helpful if used at a dose of completing at least three techniques per week. Engagement data will be captured using the RxWell PowerBI analytics dashboard. The number of completed techniques and number of messages sent to the coach will be analysed. Results of a quality improvement project using RxWell for AYAs at University of Pittsburgh Medical Center demonstrated that use of the dCBI was associated with a significant decrease in anxiety and depression scores between baseline and 1 month, and up to 3 months.16

{kind=link}
{kind=link}
RxWella screenshots: digital coaching, goal setting, CBT technique library and practice again. aThe ‘Depression Track’ programs have a library of 40 techniques, respectively. Users have access to a goal-setting tab and an ‘in the moment relief’ section which contains 17 techniques, 14 of which have brief audios to help users engage relaxation responses. CBT, cognitive behavioural therapy.
Participants in both groups (intervention and control) will receive up to $75 (USA) compensation for participation in the study.
Coaching model
RxWell includes an integrated digital health coach that is assigned to each user. Coaches are bachelor’s-level graduates who complete additional training in motivational interviewing and CBT for treating anxiety and depression. Health coaches are supervised by a licensed mental health clinician. The health coach communicates with users via asynchronous, secure, within-app messaging. Their role is to reinforce CBT principles, guide users through goal setting, motivate and help users work through challenges, recognise successes and humanise the experience for the user.
The coaches interact with their users within the app through a coaching dashboard. These interactions are based on all information that a user inputs into the app, including the messages to their coach and the techniques completed. Decisions made by the coach to modify a user’s path will be monitored during weekly coach supervisions and other quality assurance processes in place. Coaches message every 2 to 3 days to keep the user engaged or motivate them for initial engagement periods with less frequent engagement if users do not respond.
Study risk management protocol
Risk assessment for each user is completed daily, through review of all messages by the coach. Digital health coaches follow a risk escalation protocol, which includes recognising any signs of risk, from increased distress to suicidal or homicidal ideation, reaching out to the user appropriately based on the level of risk and contacting their supervisor. AYAs who appear to be at risk for harm or deterioration based on increased distress from their write-in responses in the techniques, significant increase in GAD-7 or PHQ-9 scores and/or messages to their coach receive specific action. The digital health coach will reach out to their licensed mental health supervisor about the observations. Concurrently, the coach will message the user and encourage them to reach out to their medical provider or 911, depending on the severity of escalation. Licensed supervisors determine the need for further outreach to AYAs, referring clinicians and/or pre-identified study site mental health providers.
Primary outcome measures
The primary outcome is depression severity at 12 weeks measured by the clinician-rated CDRS-R and the self-reported PHQ-9 measure. These measures will also be administered at baseline and 6 weeks after baseline. Our rationale for using both is that studies have shown that although self-rated and clinician-rated depression measures are moderately to strongly correlated at baseline, they probe different symptoms and are subjected to differences in report bias, depressive and personality subtypes and ethnic and socioeconomic factors that make the use of both valuable in measuring treatment effectiveness.17 18
Clinician-rated CDRS-R
The CDRS-R is a rater administered 17-item interview completed by phone, with item ratings between 1 (=no difficulties) and 5 or 1 and 7 (=clinically significant difficulties) (adding up to a total score between 17 to 113). A score of ≥40 indicates depressive symptomatology and a score ≤28 often indicates remission within trials.19
Patient Health Questionnaire-9
The PHQ-9 has 9 items, each scored from 0 to 3 for 27 maximum total (24 maximum total at screening in this study due to exclusion criteria). Total scores indicate depression severity. A score of 0–4 indicates no depressive symptoms, 5–9 indicates mild depressive symptoms, 10–14 indicates moderate depressive symptoms, 15–19 indicates moderate severe depressive symptoms and 20–27 indicates severe depressive symptoms.15
Secondary outcomes measures
The secondary outcomes include anxiety severity (GAD-7), evidence of passive SI (item 9, PHQ-9), general quality of life (SWLS) and CGAS score (completed by a blinded rater). All measures are completed at baseline, 6 weeks and 12 weeks. All self-reported measures will be completed online through the secure data collection platform, REDCap Cloud and the clinician-rated CGAS will be completed by phone.
Suicidal ideation
The question about suicidality is item #9 in the PHQ-9, and the score is a 0–3 range. This is a sensitive indicator of SI.20 21
Generalised Anxiety Disorder-7 (GAD-7)
The GAD-7 has 7 items and a total score that can range from 0 to 21. Total scores of 0–4 indicate no anxiety symptoms, 5–9 indicate mild GAD symptoms, 10–14 indicate moderate GAD symptoms and a score ≥15 indicates severe GAD symptoms.22
Satisfaction with Life Scale (SWLS)
The SWLS is a self-rated 5 item measure that is scored from 1 to 7 with a maximum score of 35. Higher scores correlate with higher satisfaction, with a range from extremely satisfied (scores 31–35) to extremely dissatisfied (<9).23
Children’s Global Assessment Scale (CGAS)
The CGAS is a rater-assessed measure that indicates the level of general functioning over the past month, focusing on the subject’s most impaired level of functioning during that period on a hypothetical continuum of health-illness. Scores range from 1 to 100 with 100–91 corresponding to superior functioning and 31–40 with major impairment in functioning in several areas and unable to function in one of those areas.24
Covariates
Covariates have been selected a priori based on clinical and scientific rationale: gender, race/ethnicity, parental education and insurance (public vs private). Gender and race/ethnicity will be collected from a self-report measure.
Statistical methods
Sample size and sampling design
Sample size estimate: 450 (150 per site) participants will be randomised to dCBI+TAU. Assuming 70% enrolment in the app, it is expected that n=315 will initiate the intervention (105 per site). 300 (100 per site) participants will be randomised to TAU alone. With 30% anticipated attrition at 3 months in both treatment arms, it is predicted that n=430 (220 in dCBI+TAU, 210 in TAU alone) will have complete 3-month data. Any consented patients who are excluded from randomisation and who are lost to follow-up will be recorded.
Power analysis: N=430 patients (n=220 dCBI+TAU, n=210 TAU) are expected to have complete 3-month data. However, multiple imputation will be used for intent-to-treat (ITT) analyses to retain the full n=615 (n=315 dCBI+TAU, n=300 TAU). Power is provided for both the complete data sample and the ITT sample. Based on our phase 1 open trial of dCBI in 506 AYAs with anxiety/depression, a PHQ-9 between-group Cohen’s d effect size of d=0.40 is expected for baseline-to-12-week change in depressive symptoms. With α=0.025 (two primary outcomes), >0.97 power is expected to detect this difference using a two-sided two-sample t test for samples of n=430–615.
Allocation of participants to study and control arms
Participants will be randomly assigned to one of the two treatment arms by block site randomisation with a 3:2 ratio using computer generated randomisation sequence for each site, with block size randomly selected to be 5 or 10. Randomisation does not include stratification. Approximately 33% of participants will come from each of the three sites. Research staff at each site will enrol participants.
Data collection, handling and analysis
Data will be entered into REDCap Cloud. The REDCap Cloud server offers robust security to ensure privacy and is accessible with restricted permission to study team members. A bimonthly check will occur for quality purposes. The coordinating centre will oversee the intrastudy data sharing process.
Preliminary exploratory analyses will be conducted to examine missing data and reasons for why the data are missing, assess distributions and check for outliers. Exploratory analyses will be conducted to ascertain data characteristics and screen for outliers, investigate the internal consistency and reliability of the measurement scales using Cronbach’s alpha and verify the statistical assumptions of the planned primary analyses. If assumptions are violated, alternative procedures such as data transformation or more robust statistical methods will be considered. Missing data will be examined using available data on subject characteristics. Logistic regression models will be created to compare characteristics of subjects who remained in the study versus those who dropped out. If data are determined to be missing at random, multiple imputation and/or likelihood estimation procedures will be used to produce unbiased estimates and allow retention of participants with missing data. If data are determined not to be missing at random, pattern mixture or selection modelling will be used to investigate attrition. Finally, a summary will be completed of baseline demographic and clinical information on participants and intervention usage/fidelity for each arm.
Aims 1–2: the primary outcome assessment is an intent to treat (ITT) analysis.25 Across all analyses, site effects will be examined by performing stratified analyses and examining interactions. For the primary 12-week outcomes, a linear mixed effects model will be used to regress the 12-week CDRS-R on group (dCBI+TAU vs TAU alone), baseline CDRS-R and covariates. A similar model will be fit using PHQ-9 instead of CDRS-R. Secondarily, a linear mixed effects model will be used to regress each repeatedly measured depression outcome on categorical time (baseline, 6 weeks, 12 weeks), group (dCBI+TAU vs TAU) and the time by treatment interaction. A significant interaction will indicate that the groups differ in their time course. Mixed models will include a random site effect to account for nesting. The study will plan to use comparisons to test whether changes from baseline to 6 and 12 weeks differ between groups as well as to determine whether the improvement in each group is linear. The former test will indicate group differences in slope, while the latter test will indicate whether changes are linear or non-linear. Covariates will include gender, race and socioeconomic status. For continuous secondary outcomes (anxiety, QOL, functioning), a similar modelling strategy will be used as outlined for the primary outcome. For SI, a generalised linear mixed effects model with a logit link will be used. The 6-week time point will be used to explore the slope of change of depressive severity. Finally, as an exploratory subaim, we will quantify the strength of association among the PHQ-9 and CDRS-R. To accomplish this, we will examine correlations between scales at each visit as well as the correlations between changes in each scale (eg, from baseline to 6 weeks and 6 weeks to 12 weeks). We will also use the linear mixed effects modelling strategy defined above to regress the CDRS-R on the PHQ-9.
Aim 3: in the mixed effects models described above, the study will test potential moderators (race, socioeconomic status, geography and expectancy) of the treatment effect on the outcome with a three-way interaction of treatment, time and the moderator. If significant, contrasts and stratified analyses will be used to further interpret results. If the three-way interaction is non-significant, the main effects will also be tested to identify non-specific predictors. Predictors of treatment engagement will also be examined within the dBI+dTAU group, defined as completing at least three techniques. Finally, per protocol analyses will be explored, including only participants in the treatment arm who had sufficient engagement, defined as at least three techniques for descriptive purposes. Additional app-related variables (eg, time spent using app, number of techniques repeated) will be examined. Data will be analysed using R.
Safety
Breach in participant confidentiality will be minimised by assigning a unique study ID to each participant. All data will be secured in REDCap Cloud, which has multiple layers of security as a Health Insurace Portability and Accountability Act (HIPAA) compliant research database platform. HIPPA is a US federal law that required a creation of national standards to protect sensitive health information from being disclosed without the patient’s conset or knowledge.26 The dCBI only collects a patient’s email address to send a verification code to download the secure app. The email is housed in the dCBI HIPAA compliant administrative portal that is not accessible to the coaches or study team.
Patient and public involvement
None.
Ethics and dissemination
All participants or their parent/guardian (under 18 years or unemancipated) will give informed consent to a study team member. Each participant will receive a PDF file of the informed consent form. The Institutional Review Board (IRB) is a board at each institution in the United States that reviews and monitors research involving human subjects. This study protocol was approved by the University of Pittsburgh IRB on 30 November 2021 (STUDY21080150). The study subsequently receive approval from University of California San Diego’s Human Research Protection Program IRB on 14 July 2022 (802047) and Boston Children’s Hospital IRB on 25 October 2022 (P00040987). Study results will be shared through peer review publication and presentation at national meetings plus dissemination of the findings back to providers and stakeholders.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @kathyhollenbach
Contributors The principal investigator ES who is the corresponding author of this manuscript is involved in preparation of protocol and revisions, organising scientific advisory board meetings, and publications of reports. DW and SB are involved in study oversight, and revision of the protocol and manuscript. KS-M, GJ, EHL, DRD and KH are also involved in the study oversight, and revision of the protocol and manuscript. KW, MB and DMY are involved in the revision of the protocol and manuscript. MW is involved in the revision of the protocol and manuscript as well as the statistical analysis and data summaries of the study.
Funding This work was supported by a donation from YourMomCares (no award or grant number is associated with this donation).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
© Author(s) (or their employer(s)) 2023. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.