Article Text

Abstract
Introduction Adherence to HIV antiretroviral therapy (ART) remains the cornerstone of HIV treatment. For individuals with suboptimal adherence, electronic adherence monitoring (EAM) technologies have become an important component of multimodal adherence support strategies. Most EAM technologies detect pillbox opening, and therefore, assume but cannot verify actual ingestion of oral medication. In contrast, a digital pill system (ID-capsule manufactured by etectRX, here named My/Treatment/Pill) measures directly ingestion of medications. Identifying the superior method to measure ART adherence would improve virological suppression by enabling the delivery of real-time interventions to support ART adherence, particularly in high-risk populations.
Methods and analysis Cross-over, randomised trial with 1:1 variable block size randomisation comparing two EAM systems in prescription opioid-using HIV+patient on once daily oral bictegravir, emtricitabine and tenofovir alafenamide regimens and detectable viral load >200 copies/mL within 30 days of screening (n=80). The primary outcome is once daily ART adherence measurement efficacy as assessed by comparing the accuracy of each EAM system as measured by concordance of the respective EAM systems to dried blood spot ART concentrations. Secondary outcomes are the identification of multilevel factors that are prevalent in the target population most closely linked to ART non-adherence and EAM non-adherence.
Ethics and dissemination This protocol was approved by the institutional review boards of participating sites (The Ohio State University, The Fenway Institute and the University of Miami). Data will be presented at scientific conferences and published in peer-reviewed journals.
Trial registration number NCT03978793.
- Substance misuse
- BIOTECHNOLOGY & BIOINFORMATICS
- HIV & AIDS
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
The study involves four different measures of antiretroviral adherence.
The study compares two electronic adherence technologies.
The study involves a cross-over design where participants serve as their own controls.
A limitation of the study is that participants know which electronic adherence measure they are using.
The study design will allow for future evaluations of the economic costs and impacts of the two different adherence technologies.
Introduction
Continuous adherence to HIV antiretroviral therapy (ART) among people living with HIV (PLWH) is critical to maintaining viral suppression and prevention of transmission of HIV.1 Despite the widespread availability of ART in the USA, adherence remains suboptimal; HIV-infected (HIV+) patients who take opioids have even greater difficulty adhering to ART regimens for reasons that remain unknown. Additionally, individuals who develop opioid use disorder may have additional barriers to adherence like worsening mental health, stigma around HIV and substance use and trauma that result in decreased ART adherence and less engagement with HIV care. These factors cascade into a lack of viral suppression and worsening HIV infection as well as an increased risk of transmission. Among PLWH, there is nearly three times greater incidence of opioid misuse compared with individuals without HIV. In this population, measuring ART adherence is important in order to detect individuals who may experience nonadherence and provide empiric interventions geared towards addressing HIV related comorbidities and skills-based ART adherence training.
Multiple modalities to measure ART adherence exist. These range from indirect methods like pill counts, pharmacy refills and self-report to direct measures like directly observed ingestions, ingestible sensors and video directly observed ingestions. Additionally, biological testing for components of ART like tenofovir diphosphate (for cumulative adherence) and emtricitabine triphosphate (FTC-TP) (for acute adherence) suggest average rates of adherence over time. Each strategy is marred with inconsistencies, yet with increasing ubiquity of mobile phones and access to the internet has led to a renewed interest in using electronic adherence monitoring (EAM) tools to measure and respond to ART adherence patterns. EAM has emerged as an important component of multimodal strategies to support medication adherence.2
Two EAM systems that are used to measure ART adherence are electronic pill bottles and digital pills. Electronic pill bottles (eg, Wisepill, MEMS caps) detect container opening only before sending real-time information to clinicians. These methods’ greatest weakness is that they assume but cannot verify actual ingestion of oral medication.2 3 A contrasting technology involves digital pill systems, an innovative constellation of technologies that detects actual medication ingestion. The digital pill system, known in this study as ‘My/Treatment/Pill’ (‘MyTPill’) comprises a gelatin capsule that over encapsulates several advanced technologies as well as a medication such as Biktarvy. After a patient swallows the digital pill, gastric contents dissolve the gelatin capsule in less than 60 s to release the ART tablet. Chloride ions in gastric fluid activate a tiny battery that powers a radio emitter that transmits a unique code to identify ingestion. The pill’s transmitter has a nominal operation time of 30 min, but will continue to transmit as long as remains in the stomach.
The radio signal from the digital pill is transmitted via a wearable reader that is worn using a neck lanyard (or set up as a base station) to a HIPAA-compliant app running on a smartphone. The reader acts as a store and forward device which collects ingestion data via the digital pill, retains a copy of ingestion data on its internal memory and transmits this data to a smartphone app using low energy Bluetooth. The app ‘syncs’ with a web interface so that adherence data is stored on a secure server (similar to how email on a smartphone syncs with email presented on a desktop or laptop). To prevent use by others, the app is password protected. Furthermore, the app is designed to avoid stigma while protecting confidentiality; even the icon for the app contains no image that can be construed as being related to any medication or HIV. At study completion, the app and all study data will be removed from the smartphone. The digital pill system was cleared by the U.S. Food and Drug Administration in 2019.
The digital pill system does two essential things: (1) measures if and when patients ingest ART and (2) serves as a platform for timely interventions that promote ART adherence and persistence.4 5 Digital pills, therefore, have the unique ability to provide vivid, indisputable measures of medication ingestion. No head-to-head comparisons of EAM have been performed, nor has the optimal method to incorporate the optimal EAM into clinical care been identified. Identifying the superior method to measure ART adherence would improve virological suppression by assessing, in real time, the impact of interventions to support ART adherence, particularly in high-risk populations.
In this study, the digital pill comprises a standard, commercially available 000 gelatin capsule containing a radio emitter (IDCap; eTectRx, Newberry, Florida, USA). A standard pill filling machine adds a single oral pill of bictegravir/emtricitabine/tenofovir alafenamide (Biktarvy) to each capsule creating the digital pill. Each digital pill contains its own radio emitter. Gilead Sciences will be supplying the bictegravir/emtricitabine/tenofovir alafenamide coformulated medication to be compounded in the digital pills and used in Wisepill.
Objectives
The primary aim of this investigation (R01DA0472360) is to evaluate the efficacy, usability and acceptability of MyTPill to measure ART adherence as compared with Wisepill. We hypothesise that MyTPill will provide the more accurate measure of recent and cumulative ART adherence and be reported by participants to be an equally acceptable and usable strategy to measure adherence, as compared with Wisepill.
As a secondary aim, we will examine by timeline follow back (TLFB) and qualitative interviews which aspects of prescription opioid use, misuse and abuse; pain severity; withdrawal; and demographic, social, structural and other environmental contexts are most closely linked to ART adherence. We will assess these aspects by TLFB and qualitative interviews. We will use dried blood spot (DBS) concentration ART assays as the ‘gold standard’ of ART adherence in clinical trials.6–10 Furthermore, we will examine how these aspects affect changes in ART adherence as assessed by MyTPill and Wisepill.
Trial design
To assess the primary aim, this randomised trial will use a 1:1 variable block size randomised cross-over design to assign prescription opioid-using HIV+ patients taking onc daily ART regimens and viral load of >200 copies/mL to one of two arms: (1) MyTPill × 3 months, then Wisepill × 3 months or (2) Wisepill × 3 months, then MyTPill × 3 months. The study is a cross-over design that allows for same-group comparisons through two different interventions (MyTPill or Wisepill). Because of this design, a cohort’s responses to the intervention, in regard to adherence and perceptions toward the interventions themselves, can be compared. In addition, any effect due to the order of receipt of the intervention can be compared across groups.
Methods and analysis
Study setting and recruitment
Participants will be recruited from the Jackson Memorial Hospital HIV Clinic in Miami-Dade County, Florida and Fenway Health in Boston, Massachusetts. Recruitment is facilitated by registries and on-site research staff who can screen for study participation and collect and process biological specimens to screen for prescription opioid use. The Jackson HIV Clinic provides care for >250 HIV+ patients maintained on oral prescription opioids for chronic pain; Fenway Health delivers care to approximately 300 such patients. At both sites, approximately 30% of HIV+ patients on opioids have suboptimal adherence, although the number of HIV+ persons taking prescription opioids is increasing annually. Because relying extensively on recruitment from the Jackson HIV Clinic could limit enrolling a diverse cohort of HIV+ opioid users, community-based recruitment efforts targeting the study population (eg, flyers, face-to-face recruitment, print advertisements) will be employed in addition to clinic advertisements and clinician referrals should accrual lag.
Participant recruitment, enrolment and retention will be summarised using the CONSORT guidelines.11
Eligibility criteria
Trained research staff at each study site will recruit patients who are prescribed an ART regimen containing bictegravir, emtricitabine and tenofovir alafenamide, have a detectable HIV viral load (>200 copies/mL) at the time of screening, and take prescription opioids including buprenorphine and methadone. We will collaborate with clinic staff to obtain referrals for potentially eligible participants to study staff who will conduct formal screening to confirm inclusion criteria. The medical record will be queried to confirm ART regimen and prescription opioid use. At an in-person screening visit, participants will additionally provide urine, which we will assess for the qualitative presence of opioids to confirm opioid use. For women of childbearing age, we will additionally obtain a urine pregnancy test to confirm they are nonpregnant. We will review the medical record for the following exclusion criteria: hypersensitivity to silver, magnesium or zinc following oral use; chronic liver or renal disease (estimated creatinine clearance <30 mL/min); active pregnancy as detected on a urine pregnancy test; non-English speaking; history of Crohn’s disease or ulcerative colitis; history of bowel surgery, gastric bypass or bowel stricture; and history of gastrointestinal malignancy or radiation to the abdomen.
Patient and public involvement
No patients participated in protocol development.
Study methods
The patient registry at participating sites will be reviewed by treating clinicians for potential participants; these individuals will be screened for participation by study staff in a private office from February 2021 to May 2023. Potential participants then return for an enrolment visit where the study objectives, respective EAMs, risks and benefits are reviewed. Participants are enrolled only after providing written informed consent (see online supplemental file). When a new participant completes the screening and enrolment visit, the study physician will prescribe a 30-day supply of bictegravir/emtricitabine/tenofovir alafenamide for both the MyTPill arm (formulated as a digital pill) and the Wise Pill arm. The RA will then fax the prescription for the study medication to an outside specialty pharmacy, ARx Patient Solutions Pharmacy, that will prepare the investigational product (IP) and package the capsules in containers labelled as appropriate for the patient and study. The study medication will then be shipped to the research pharmacy at the study site. This compounded ART medication will be provided to the patients at no cost. The study drug will be shipped from Gilead to ARx Patient Solutions Pharmacy in bulk. An RA will demonstrate proper use of the EAM technology to the patient at time of intervention initiation and will contact the participant in the event of malfunctioning or missing devices.
Supplemental material
The eighty participants are to be assigned using 1:1 variable block size randomisation into one of either (1) the MyTPill first, then Wisepill arm or (2) the Wisepill first, then MyTPill arm (figure 1). Electronic randomisation, stratified by site, occurs at the point of informed consent. Each study period (ie, MyTPill arm or Wisepill arm) will last 3 months, followed by a 2-week wash-out period when they will use no adherence monitoring system (6.5 months total). Every participant will receive a 2-week supply of study drug before they started the wash-out period. In the absence of any data guiding the optimal length, a 2-week wash-out period was deemed practical and feasible so to allow for changing of equipment and maintaining participant engagement. Within the first 2 days of starting the study, research staff will contact participants to confirm correct technology use and answer any questions.
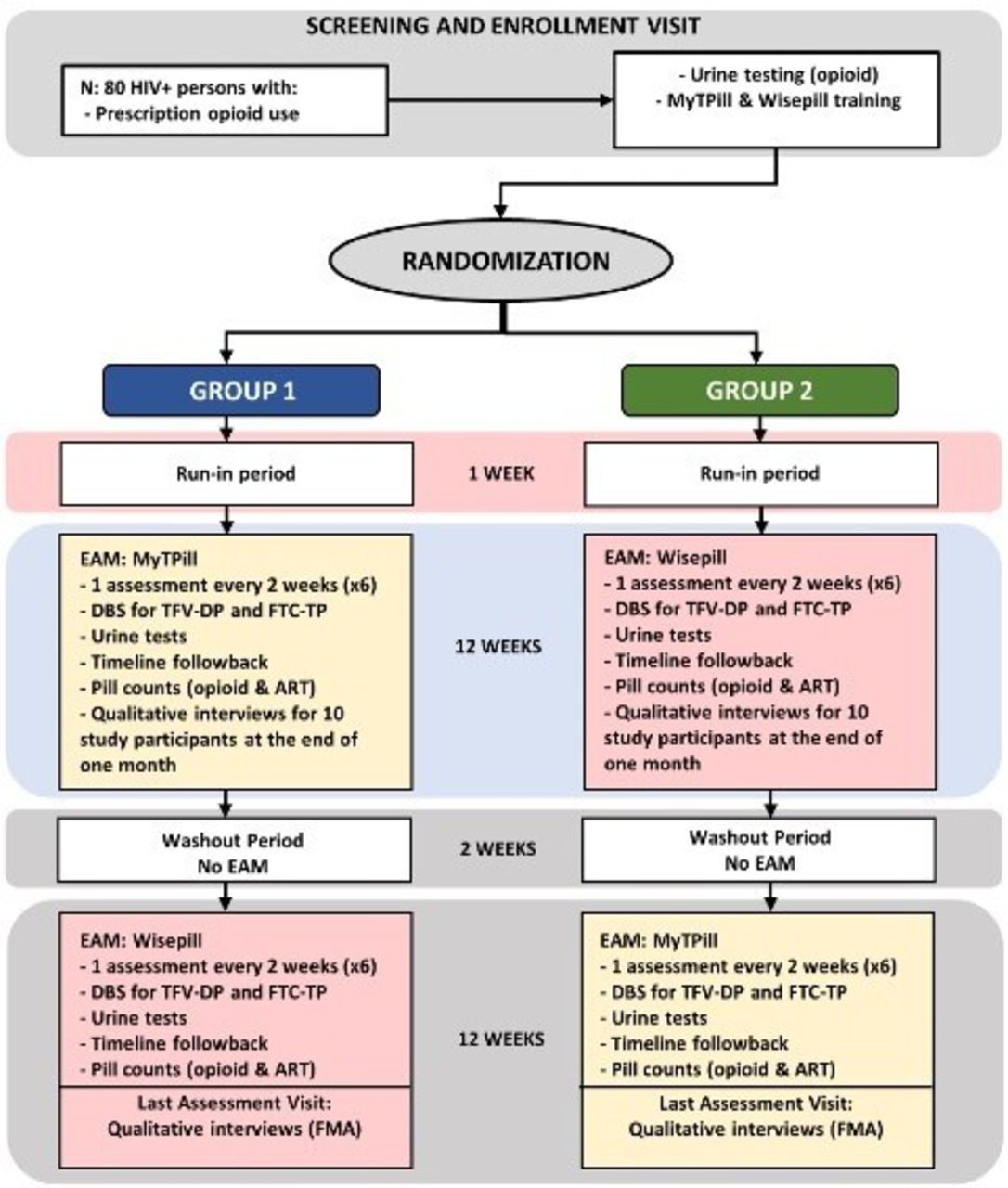
{kind=link}
Study activities. ART, antiretroviral therapy; DBS, dried blood spot; EAM, electronic adherence monitoring; FMA, framework matrix analysis; FTC-TP, emtricitabine triphosphate; TFV-DP, tenofovir diphosphate.
Participants in both study arms will be given a 1-week run-in period with the technology that they are randomly assigned to, after enrolling in the research study. A digital encounter will be scheduled to ensure that the participants are comfortable in making the 6-month commitment to incorporating EAM technology in their daily lives. The study participants who were unable to successfully complete the run-in period or are unwilling to continue, will be excluded from the study and the reasons for their inability to complete the run-in period will be assessed through a qualitative interview. After completing 3 months with the assigned technology, the participants will not be subjected to an additional run-in period with the second technology as we presume that those who accept an EAM will accept both of our form factors. It is unlikely that participants would need to stop their ART due to adverse events since they have already been taking the medication before the trial began. However, ART stoppage not related to the trial would be considered similar to drop-out of the study. In the unlikely circumstance, the participant would be disenrolled and all data will be censored at the time of drop-out. All study activities will occur in addition to subjects’ routine clinical care.
Follow-up assessments
Participants will complete up to 15 follow-up assessments over the 6-month investigation (tables 1 and 2). They will be asked to present to the clinic from which they were recruited on a scheduled day every 2 weeks (±3 days) during the 24 weeks when their ART medications are being monitored (a total of 13 visits). Study visits 5, 7, 9, 12, 14 and 16 will be conducted partially remotely and partially on site. While visits 4, 6, 8, 10, 11, 13 and 15 will be conducted entirely in person either for technology demonstration to subjects or for biological samples collection.
Schedule of activities: treatment and follow-up period—the first 3 months
Schedule of activities: treatment and follow-up period—the final 3 months
Follow-up visits will be conducted every 2 weeks and reminder notices will occur prior to their appointment via text or phone call. Prior to the in-person portion of this visit, we will review the previous 30 days of adherence data over using a virtual video conferencing system (eg, Zoom) and reconcile discordant or nonadherent events with participants in both study arms, confirm correct usage of the technology and obtain the self-reported last ART dose. During the in-person portion of this visit, participants will return any unused pills from the prior 30-day period and perform pill counts for both opioids and ART by study staff. We will perform phlebotomy to obtain a single tube of blood which we will use to perform DBS testing for tenofovir and emtricitabine-based measures of adherence (and to compare DBS against the MyTPill and Wise Pill), and a urine drugs of abuse immunoassay assessing opioid substance use. DBS test will be conducted in all study visits except visits 1, 2, 3 and visit 10. Inventory of IP and any returned product that is collected will be maintained for drug accountability/pill counts.
We will conduct TLFB interviews for event-level associations related to prescription opioid use, pain, ART non-adherence, affect and substance use. TLFB has demonstrated good test–retest reliability, convergent validity and agreement with collateral reports for substance use and ART. The TLFB interviews are designed to collect information regarding event-level prescription opioid use (including opioid ingestion/dose/frequency), ART adherence, pain severity, withdrawal, use of other substances and mood in the previous 30 days that we have previously implemented. The TLFB first reviews critical life events retrospectively to prompt recall of data that are recorded on a personalised calendar.
In addition to the TLFB interviews, we will conduct qualitative semistructured interviews with each participant after 3 and 6 months of enrolment to assess EAM usability and acceptability. Interviews will be conducted with all participants, including those who choose to drop out of the study prior to completion of all study activities. Interviews with those participants will assess their reason for study cessation, interviews with all participants will seek to understand their day-to-day experiences with MyTPill and Wisepill; acceptability of each system, including how the user experience of the systems can be improved; and whether they felt system use impacted their adherence.
Outcomes
The primary outcome of ART adherence measurement efficacy will be assessed by comparing the accuracy of the MyTPill and Wisepill and will be assessed as concordance of these two respective EAM systems versus DBS samples results. Secondary outcomes are the identification of multilevel factors that are prevalent in the target population most closely linked to ART non-adherence and EAM non-adherence.
Sample size
Our sample size of 80 participants (40 per study arm × 2 study arms; estimated 20% attrition) is based on satisfying the primary aim of the study: the comparison of ART adherence efficacy of the MyTPill versus the Wisepill system. The benefit of the cross-over design permits comparison of participants across study arms (MyTPill first 3 months vs Wisepill first 3 months and MyTPill second 3 months vs Wisepill second 3 months) and within study arms (MyTPill vs Wisepill for the same participant). Comparisons within study arms permit a reduction in variance and enables detection of statistical difference with smaller effect sizes, as compared with the across study arms comparisons.
The accuracy of the MyTPill and Wisepill measurements will be determined by examining the concordance between these measurements and the ‘gold standard’ DBS. As such, concordance can be considered a continuous value with a range from 0 to 100. Recent adherence is assessed by the presence of FTC-TP in DBS samples in a binomial fashion: adherent or non-adherent within the prior 48 hours. In an analogous fashion, MyTPill and Wisepill indicate whether or not ARTs were ingested (MyTPill) or the pill bottle opened (Wisepill) at least once in the prior 48 hours. Two DBS will be obtained per participant per month (12 total; 6 DBSs during Wisepill and 6 DBSs during MyTPill) for a total of 960 DBSs. If 20% are not collected or are not readable for any reason, there would be 768 samples. For this investigation, which is the first of its kind, there is no clinical standard to base the effect size, and no prior pilot studies to base the estimate. An effect size of 10%–15% seems to be a reasonable gestalt estimate in comparing two behavioural interventions for adherence. Using an estimate absolute difference of 10% reflects a hypothesis that one intervention has 10% greater absolute adherence than the other intervention. The sample size is based on the number of blood samples that are concordant and as such a sample size of 80 participants providing at least 768 dried blood samples permits approximately 90% power in detecting that absolute difference. The study would have greater power for a larger effect size of 15%, if detected. In this case, the comparison is two-sample binomial proportion. Using Fisher’s exact test to examine differences in measures of concordance between the MyTPill and Wisepill, we can detect with high power (>0.9) an effect size of an absolute difference of at least 10%–15% of greater concordance with the DBS gold standard.
Statistical methods
Participant demographic information will be summarised using conventional statistics as stratified by study arm. Baseline characteristics will be compared using Pearson’s χ2 test of Fisher’s exact test for categorical variables and Student’s t-test for normally distributed or Wilcoxon’s test for non-normally distributed continuous variables to assess the success of the randomisation procedure. Patterns in missing data will be examined and imputation performed if necessary. The technique for handling missing data, if it occurs, will depend on the type of missing data. If possible, multiple imputation using available covariates will be conducted.
Any chance imbalances between the two arms will be documented and investigated. If possible, these imbalances will be adjusted for by including the imbalanced covariate as an independent variable in our subsequent analyses. A two-tailed, α=0.05 level of significance will be used for the analyses.
Three assessments of ART adherence measurement efficacy for both recent and cumulative adherence will be conducted. First, we will compare concordance between MyTPill and Wisepill and the DBS gold standard throughout across the entire study period by calculating a Pearson’s correlation coefficient. We will consider adherence on a continuous scale based on percent adherence detected from the EAM and compare this with projected adherence as described through DBS. Second, we will test if there is an effect by study period because of changes in behaviour over time, due to follow-up assessments, and clinic visits (during which clinicians might counsel participants on improving ART adherence, which may affect concordance with DBS measurement). We will compare concordance across study arms during the first 3-month period and we will compare 3-month concordance rates across study arms during the second 3-month adherence assessment period (ie, assigned to MyTPill for the second 3 months vs Wisepill for the second 3 months). Third, we will compare concordance with DBS ART adherence measures within study arms between the first and second 3-month adherence assessment periods (eg, assigned to MyTPill for the first 3 months vs Wisepill for the first 3 months). For all comparisons, we will calculate mean concordance by study arm, then perform two-sample tests of binomial proportions (adjusting for multiple measurements per person, as required), and estimate the differences between study arms along with corresponding 95% CIs, taking into account period and carryover effects if necessary.
In addition, we will calculate test performance characteristics (sensitivity, specificity, predictive values and likelihood ratios) of MyTPill and Wisepill versus DBS for each ART adherence assessment period (full 6 months, first 3 months, second 3 months) along with corresponding 95% CIs.
To assess patient experience (usability and acceptability), qualitative interviews with participants at the end of each 3-month study period will be conducted, audiorecorded, transcribed verbatim and analysed using a framework matrix analysis, a common qualitative data reduction technique ideal for practice-oriented findings.12 13 NVivo V.20 qualitative data analysis software, which contains a framework analyses tool, will be used to review, summarise and classify data. Extensive analytical memos will be written after each interview is conducted, coded and throughout the analysis process to reflect on code choices, emergent themes and patterns, and conceptual models. Individual participant comments about key content are entered into the software; the comments are then reviewed in aggregate to identify themes and trends in user experiences. Finally, the data will be themed, in which the final sets of codes and their meanings will be transformed into more descriptive themes to organise recurrent meanings.
The analyses for the secondary aim focus on identifying factors associated with ART nonadherence, as measured by DBS alone, and factors associated with ART adherence as measured by MyTPill and Wisepill. In addition, we will measure factors associated with ART adherence measurement concordance as assessed by DBS versus MyTPill or Wisepill, respectively. We will begin with examining factors associated with greater ART adherence collapsed across study arms for the first time adherence is measured (either recent or cumulative); since recent adherence is a binary variable and cumulative adherence is an ordinal variable as measured by DBS, we will create univariate logistic and ordinal logistic regression models using DBS-measured ART adherence as the outcome, and in an exploratory fashion examine factors associated with greater or lesser adherence individually. Factors considered will be study arm, demographic characteristics, self-reported ART adherence, pill count-measured adherence and information collected during the TLFB and other study assessment, including prescription opioid use (including opioid ingestion/dose/frequency); pain severity; withdrawal; use of other substances; mood and demographic, social, structural and other environmental contexts. We will then use these exploratory analyses to guide the creation of multivariable models. Factors that are associated statistically with ART adherence at least at the p<0.10 level in our exploratory analyses and variables found to be associated with ART adherence in prior research will be considered further in multivariable model construction.
Using this model as a general approach, we will repeat the model building exercise for the first time ART adherence as measured by MyTPill and Wisepill, respectively; since we expect that we will be able to measure adherence by both MyTPill and Wisepill as continuous variables, we will examine the adherence distribution and choose an appropriate model based on distribution: linear if normally distributed, binary logistic dichotomised at 100% adherence and <100% adherence or hurdle/zero-inflated negative binomial for simultaneous modelling of 100% adherence and <100% adherence. Afterwards, if sample size permits, we will create similar models for ART adherence measurement discordance (MyTPill vs DBS, Wisepill vs DBS), again choosing based model type on the observed distribution. Using the lessons learnt from these models, we will develop more sophisticated models taking into account the multiple adherence measurements obtained over time in each study arm. The TFLB offers a unique opportunity to gather measurements that change over time. We will use these time varying data in generalised linear models with mixed random effects or generalised method of moment approaches, both of which take into account correlation between measurements in longitudinal data. The within-participant correlations will be accounted for according to the type of analysis performed. The adjustments will be necessary when the same cohort is compared against itself as part of the cross-over design.
Patient safety
We anticipate the major risks from participation are from the potential for exposure to metal components in the digital pill, and retention of the radiofrequency emitter portion of the digital pill. Major psychological discomforts could occur during study interviews when participants are asked about their adherence patterns. Participants will be monitored for the occurrence of undesirable/adverse events. Unexpected and related adverse events will be reported to the institutional review board (IRB) within five working days of the investigator first becoming aware of the problem.
Data monitoring
A data safety monitoring board (DSMB) comprised three researchers external to the study with expertise in ART, stimulant use, technology security and behavioural interventions, will oversee the conduct of this research. The DSMB will meet at initiation, annually thereafter and at the completion of 50% participant accrual. The DSMB will review the protocol, study plan and all study documents; evaluate the enrolment and data collection progress; review human subject’s protection and data safety; make recommendations regarding continuation, termination or modification of the study plan; and provide written annual reports to the investigators and the National Institutes of Health if required. The investigators will report DSMB activities to the IRB and National Institute on Drug Abuse in annual progress reports.
Limitations
There exists a strong likelihood that MyTPill and Wisepill increases our participants’ awareness that their ART adherence behaviour is being monitored, potentially improving it (Hawthorne effect). We considered examining for a potential Hawthorne effect by cross-over randomised controlled trial design in which participants were randomised, for example, to receive: (1) MyTPill immediately or (2) MyTPill after a 3-month delay following enrolment in the trial, using viral suppression at each month as the endpoint. However, both groups in this arrangement still are likely to be equally affected by the Hawthorne effect, and any differential Hawthorne effects will be counterbalanced by randomisation.
Ethics and dissemination
Consent
Trained research assistants will introduce and discuss the trial with potential participants. The consent process will occur either in person or electronically via Zoom depending on study site and the participant’s recruitment process. Participants will provide either electronic or in person signatures as appropriate.
Data management and confidentiality
Digital Pill data are collected on a cloud-based server hosted by our collaborator etectRx. Deidentified data will be visible to study staff on a secure, password-protected web app. We will receive raw ingestion data weekly from etectRx. These data will be stored on secure computers at participating sites.
Participants will be interviewed in a secure conference room on two digital voice recorders at the study sites. To minimise the time spent with participants in the same room, the study staff will run the interviews from a different room on the study site. Participants could also be interviewed remotely (not on-site). Participants will be instructed to not say identifying information during interviews. All interview data will be under participants’ study identifier. Basic demographic information and qualitative interview data will be collected using RedCap through secure computers regulated by the study sites. The audio recording of qualitative interviews will be downloaded to secure servers at Fenway and University of Miami. These will be sent to a HIPAA-compliant transcription company (Landmark Transcription) via secure file transfer. Once we verify that the transcriptions of the recordings are complete, original audio files will be deleted from all locations (Fenway Health and University of Miami computers and voice recorders).
Access to data
The IRBs of participating sites have approved this protocol and have access to anonymised data for review as needed. No third-party investigators will have access to study data prior to planned dissemination described below.
Disseminations policy
This trial has been registered on ClinicalTrials.gov: NCT03978793. Results will be published there and in peer-reviewed journals when available by the primary study team.
Summary
This protocol has considerable public health significance. First, MyTPill will serve as a platform for subsequent research testing ART adherence interventions to address ART adherence and opioid misuse to be delivered at the moment of greatest need. Second, we will identify multilevel factors associated with suboptimal ART adherence and patterns of opioid misuse that will directly inform novel interventions for HIV+persons on prescription opioids, a growing high-risk population with 46% faster mortality. Third, MyTPill directly addresses the national crisis of opioid misuse arising from treatment of pain as a potentially viable new tool to monitor opioid misuse and diversion in various populations, a known urgent need to stem the opioid epidemic.
Ethics statements
Patient consent for publication
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @BischofMD
Contributors EWB and AWC conceived of this study; EWB, AWC, PC, YM, RP, RCM and MV contributed to the conduct of the study. RR oversaw qualitative analysis aspects of the work. JJB and CCR-G assisted with drafting and editing of this manuscript. All authors reviewed and approved final manuscript.
Funding This work was supported by the National Institute on Drug Abuse (North Bethesda, MD) grant number 1RO1DA047236-01A1.
Disclaimer Study funder had no input in study design or manuscript creation.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.