Article Text

Abstract
Objectives To assess the effectiveness and safety of insulin glargine and lixisenatide (iGlarLixi) fixed-ratio combination on a cohort of Romanian adults with type 2 diabetes (T2D).
Design Open-label, 24-week, prospective cohort study.
Setting 65 secondary care diabetes centres in Romania.
Participants The study included 901 adults with T2D suboptimally controlled with previous oral antidiabetic drugs (OADs)±basal insulin (BI) who initiated treatment with iGlarLixi upon the decision of the investigator. Major exclusion criteria were iGlarLixi contraindications and refusal to participate. 876 subjects received at least one dose of iGlarLixi (intention-to-treat/safety population).
Primary and secondary outcome measures The primary endpoint was change in glycated haemoglobin (HbA1c) from baseline to week 24 in the modified intention-to-treat population (study participants with HbA1c available at baseline and week 24). Secondary efficacy outcomes were percentage of participants reaching HbA1c targets and change in fasting plasma glucose (FPG).
Results Mean baseline HbA1c was 9.2% (SD 1.4) and FPG was 10.8 mmol/L (2.9). Mean HbA1c change was −1.3% (95% CI: −1.4% to −1.2%, p<0.0001) at week 24. HbA1c levels ≤6.5%, <7% and<7.5% at week 24 were achieved by 72 (8.9%), 183 (22.6%) and 342 (42.3%) participants, respectively. Mean FPG change was −3.1 mmol/L (95% CI: −3.3 to −2.8, p<0.001) at week 24. Mean body weight change was −1.6 kg (95% CI: −1.9 to −1.3, p<0.001) at 24 weeks. Mean iGlarLixi dose increased from 19.5 U (SD 7.7) and 30.1 U (10.0) to 30.2 U (8.9) (ratio 2/1 pen) and 45.0 U (11.6) (ratio 3/1 pen). Adverse events (AEs) were reported by 43 (4.9%) participants (18 (2.1%) gastrointestinal) with 4 (0.5%) reporting serious AEs. 13 (1.5%) participants reported at least one event of symptomatic hypoglycaemia, with one episode of severe hypoglycaemia reported.
Conclusions In a real-world setting, 24-week treatment with iGlarLixi provided a significant reduction of HbA1c with body weight loss and low hypoglycaemia risk in T2D suboptimally controlled with OADs±BI treatment.
- general diabetes
- diabetes & endocrinology
- clinical pharmacology
Data availability statement
No data are available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The main strength of the study is the large sample size, which, to our knowledge, is the largest study population for a real-world study of insulin glargine and lixisenatide (iGlarLixi) reported to date.
Another strength is that this study provides information on the glycaemic impact of the time of iGlarLixi injection (breakfast, lunch, dinner or varied timing during the trial).
In this real-world evidence study, selection bias cannot be ruled out since inclusion of patients was at the sole decision of the prescribing physician.
The study did not include participants previously treated with other glucagon-like peptide-1 receptor agonists, and sodium-glucose co-transporter-2 inhibitors were discontinued at the time of iGlarLixi initiation.
Self-reporting of hypoglycaemic episodes without the use of paper or electronic hypoglycaemia logs might have underestimated the frequency of hypoglycaemic episodes.
Introduction
Type 2 diabetes (T2D) is a progressive disease characterised by a continuous decline of beta-cell function, frequently on a background of insulin resistance.1 Due to its high and increasing prevalence and prevalent chronic complications, T2D represents a major cause of global morbidity and mortality.2 3 Landmark studies conclusively proved that good glycaemic control is associated with decreased risk of chronic complications,4 5 the benefits being maintained in the long term.6 Unfortunately, generally less than 50% of subjects with T2D reach recommended glycated haemoglobin (HbA1c) targets, both in developed and developing countries.7 8 This is true despite the major advances in diabetes management, including the development of innovative and effective medications proven to improve diabetes control in randomised controlled trials (RCTs).9
Basal insulin (BI) is a highly effective treatment in reducing fasting blood glucose.10 However, insulin treatment has the caveats of hypoglycaemia and weight gain risks, partially mitigated for the second generation of BIs.11 Because of these clinical concerns, BI initiation and intensification are often delayed by prescribers.12 On the other hand, the other injectable diabetes medication—glucagon-like peptide-1 receptor agonists (GLP-1 RAs)—is equally effective in terms of glycaemic control (both fasting and postprandial) and has the additional benefits of low risk of hypoglycaemia, weight loss and cardiovascular protection.13 However, their use may cause gastrointestinal (GI) side effects, especially nausea, which is dose related and may be prevented by slow dose escalation.14 GI side effects might partially explain the differences in glucose-lowering efficacy reported between RCTs and real-world evidence (RWE) studies with GLP-1 RAs.15
Appropriate treatment intensification with the combination of GLP-1 RAs and BI is not always seen, despite its potential benefits and its consideration in the guidelines of several scientific societies, mostly if actual HbA1c remains 1.5%–2% higher than the individualised target.9 10 Fixed-ratio combination (FRC) products that require only one injection per day have been developed, providing a simpler approach than the separate administration of either component. They have a robust glucose-lowering efficacy, similar hypoglycaemia risk but improved weight benefit compared with BI and reduced frequency of GI side effects compared with GLP-1 RAs.16 iGlarLixi represents the combination between insulin glargine 100 U/mL and the GLP-1 RA lixisenatide in a titratable co-formulation, with a fixed ratio (2/1: 2 U insulin glargine/1 µg lixisenatide or 3/1: 3 U insulin glargine/1 µg lixisenatide).17 18 The efficacy and safety of iGlarLixi were firmly established in the LixiLan clinical development programme. These trials included subjects with T2D inadequately controlled with metformin with or without another oral glucose-lowering medication–LixiLan-O trial19; subjects with T2D suboptimally controlled with BI plus metformin with or without other oral medication–LixiLan-L trial20; and subjects with T2D suboptimally controlled with previous daily or weekly GLP-1 RA with or without oral medication–LixiLan-G trial.21 In these trials, iGlarLixi proved to have superior glycaemic efficacy compared with BI glargine (LixiLan-O and LixiLan-L trials), lixisenatide (LixiLan-O trial), previous GLP-1 RA (LixiLan-G trial) and, more recently, premix insulin (SoliMix trial).22
However, data provided by RCTs usually include highly selected study populations and exclude older populations, with different diabetes and cardiovascular complications, and cannot be representative of the heterogeneous population encountered in routine clinical practice. Consequently, there is an increasing demand for data from RWE studies.23 RWE data complement those of RCTs and are essential for the assessment of the effectiveness, safety, and cost/efficiency of a particular medication within a large population of subjects with T2D. In the last years, there was increased demand from healthcare regulatory institutions for RWE studies to assess the efficiency of medicine use and the costs associated with different treatments.
Currently, there is a relative lack of RWE data regarding the use of iGlarLixi in the general population with T2D, the few available published studies being performed in Europe. Therefore, in this study, we aimed to determine the effectiveness and safety of iGlarLixi in the current clinical practice in Romania, a country with a high prevalence of diabetes at 11.7% and an increased rate of chronic diabetic complications.24 25
Methods
Study design and population
STAR.Ro was a multicentre, non-interventional, open-label, 24-week, prospective cohort study assessing the effectiveness and safety of iGlarLixi in a real-life practice setting in Romania. The study design included an enrolment/baseline visit and two additional visits: at 12 weeks±2 weeks and at 24 weeks±2 weeks.
The investigators were diabetologists, as they are the only physicians who can prescribe iGlarLixi in Romania. The study centres were distributed in all the areas of the country to ensure a good representation of adults with T2D. Study sites were selected through a randomisation process—out of the approximately 400 centres in Romania in which people with T2D are treated, 75 centres were selected randomly by a blind observer.
The sample size was calculated based on results from previous iGlarLixi trials under the hypothesis that the overall mean change of HbA1c at week 24 in patients treated with iGlarLixi should be at least −0.4% for the selected sample. To obtain an absolute precision of 0.08% with an SD of 1.1 for the change in the value of HbA1c from baseline to the end of study, 727 evaluable patients were deemed necessary. By estimating a discontinuation rate of 10%, the number of patients included was calculated to be at least 800. A target recruitment of 900 patients was planned in approximately 75 centres, with 25±5 patients/centre, but finally only 65 of the 75 centres did include patients. Each investigator included consecutive patients who met the inclusion criteria at the date of the visit and signed the informed consent form. Inclusion was capped for each site to the allotted number of patients.
The manuscript has been prepared in accordance with Strengthening the Reporting of Observational Studies in Epidemiology recommendations.26
Inclusion and exclusion criteria
Consecutive adults diagnosed with T2D for at least 1 year were eligible if they were suboptimally controlled (individualised HbA1c target) with previous therapy (at least 3 months with metformin±a second oral antidiabetic drug (OAD; either sulfonylureas, glinides, sodium/glucose co-transporter-2 inhibitors (SGLT-2i) or dipeptidyl peptidase-4 inhibitors or at least 24 weeks with metformin and BI with a dose ranging between 15 and 50 IU/day), initiated with iGlarLixi upon the decision of the investigator, with an available measurement of HbA1c during the last month prior to study inclusion.
In the study, both available iGlarLixi prefilled (Solostar) pens were used. The iGlarLixi 100 U/mL glargine+50 µg lixisenatide/mL (2/1 ratio pen) contains for each dose step 1 U of insulin glargine and 0.5 µg lixisenatide. Maximum dose that can be delivered with one injection is 40 U of insulin glargine and 20 µg lixisenatide. The iGlarLixi 100 U/mL glargine+50 µg lixisenatide/mL (3/1 ratio pen) contains for each dose step 1 U of insulin glargine and 0.33 µg lixisenatide. Maximum dose that can be delivered with one injection is 60 U of insulin glargine and 20 µg lixisenatide.
As per summary of product characteristics (SmPC) for iGlarLixi valid at the time of study design and reimbursement regulations in Romania, all OADs except for metformin were stopped at the time of iGlarLixi initiation.
Major exclusion criteria were iGlarLixi contraindications according to the SmPC and subjects who refused to sign the informed consent form.
Study outcomes
The primary endpoint was change in HbA1c from baseline to week 24 in the modified intention-to-treat (mITT) population (which included study participants with HbA1c available for both baseline and week 24 visits). Any available measurement of HbA1c during the last month prior to study inclusion was considered as baseline HbA1c.
Secondary efficacy outcomes were percentage of subjects reaching HbA1c levels ≤6.5%, <7%, <7.5% or the individualised target of HbA1c after 24 weeks and change in fasting plasma glucose (FPG). Investigators were asked to set and report an individualised target of HbA1c at visit 1 for each subject.
Other variables included gender, age, duration of diabetes, body weight, cardiovascular risk factors, chronic diabetic complications, non-diabetes concomitant medications, diabetes medication at baseline, dose of iGlarLixi at each visit, type of pen used (2/1 or 3/1 ratio) and type of titration algorithm used. Four titration algorithms were predefined:
Algorithm A: adjustment of iGlarLixi according to the median of fasting self-monitoring plasma glucose (SMPG) values from the last three measurements (>7.8 mmol/L: +4 U; >6.1–≤7.8 mmol/L: +2 U; glycaemic target, 4.4–6.1 mmol/L, inclusive: no change; ≥3.3–<4.4 mmol/L: −2 U; <3.3 mmol/L or occurrence of ≥2 symptomatic hypoglycaemic episodes or 1 severe hypoglycaemic episode (requiring assistance) documented in the preceding week: −2 to −4 U, at the discretion of the physician).
Algorithm B: +2 U every 3 days until the FPG target was reached.
Algorithm C: +1 U/week until targeted FPG was reached.
Algorithm D: any other algorithm different to the ones previously described.
For algorithms B, C and D, dose of iGlarLixi was decreased at investigator decision in case of blood glucose (BG) values <3.3 mmol/L or occurrence of symptomatic hypoglycaemic episodes or severe hypoglycaemic episodes.
Safety parameters
The hypoglycaemic events were reported in numbers, percentage and rate of events per participant per year (PPY), considering any symptomatic hypoglycaemic event (either confirmed with glycaemia <3.9 mmol/L or <3 mmol/L or unconfirmed by measurement of glycaemia) and any severe hypoglycaemic event (an event which required the assistance of another person for the administration of carbohydrates, glucagon or other resuscitative measures if the patient cannot help himself/herself).
In addition, information was collected on adverse events (AEs), serious AEs (SAEs), AEs of special interest (including GI AEs, injection site reactions/hypersensitivity reactions, medication errors and quality issues of the iGlarLixi prefilled pens).
Statistical analysis
Categorical variables were expressed as absolute numbers and percentages. Continuous variables with normal distribution were expressed as mean, SD and 95% CI; for non-normal distributed variables, median and IQR were used. Changes of HbA1c, FPG and body weight between visits were tested with paired t-test and differences between subgroups were tested with independent t-test (for two subgroups) and analysis of variance with Fisher’s least significant difference test (for >2 subgroups) using SPSS 22.0. A two-sided 0.05 significance level was applied to all tests.
Safety data were analysed using descriptive statistics.
Patient and public involvement
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research.
Results
Study population and baseline characteristics
Between 22 January 2019 and 24 December 2019, a total of 901 subjects were included in 65 study sites. Out of these, 3 were duplicates and 22 did not meet eligibility criteria, so only 876 subjects received at least one dose of iGlarLixi, defining the ITT/safety population. At the end of the study, 808 subjects had an available HbA1c value both at baseline and after 24 weeks of iGlarLixi treatment and were analysed as mITT population, while 800 were included in the per-protocol (PP) population (figure 1). The efficacy endpoints were assessed on the mITT population, except for the FPG that was evaluated on the PP population.
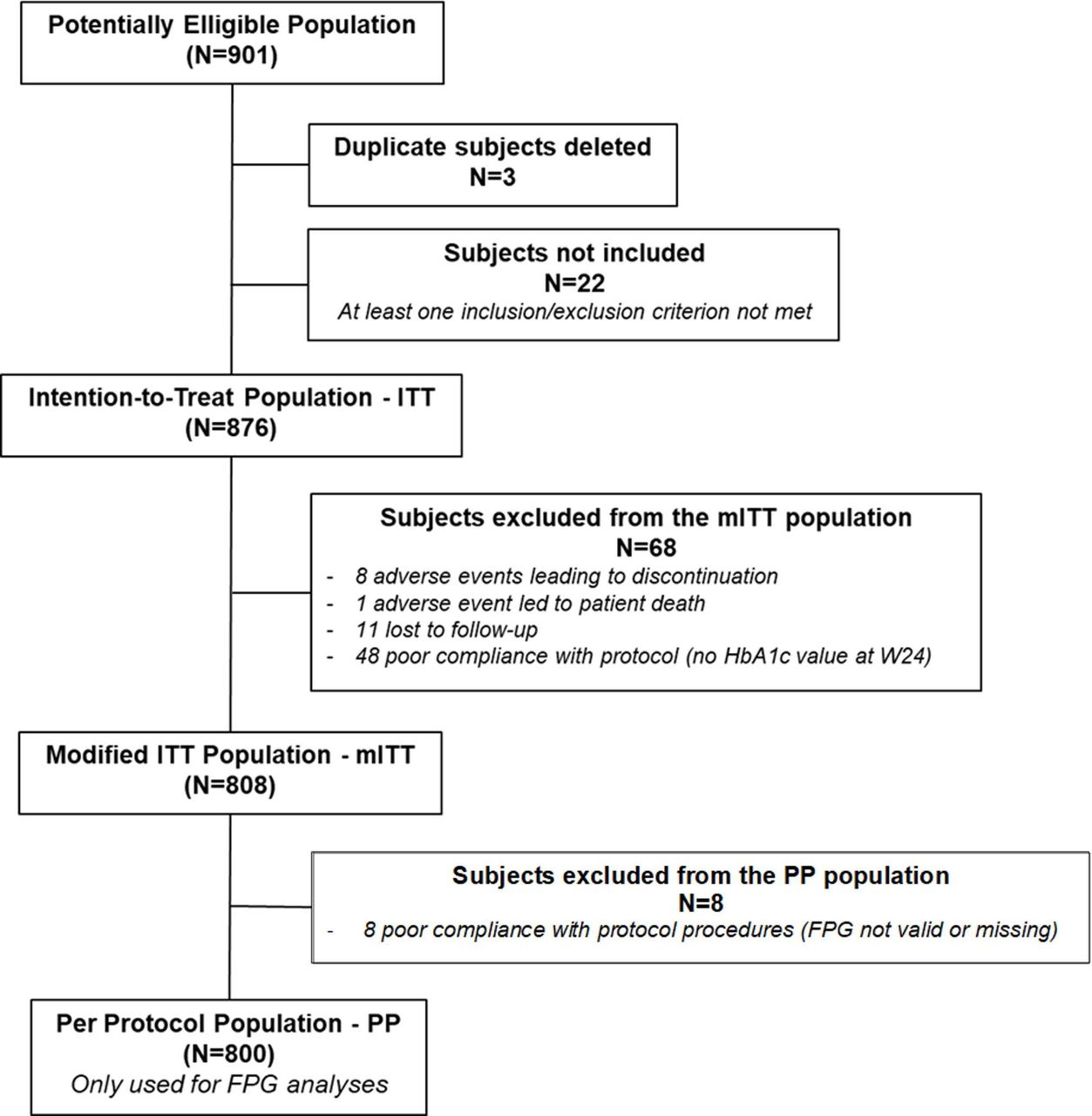
Patient disposition. FPG, fasting plasma glucose; HbA1c, glycated haemoglobin.
Demographic and baseline characteristics are reported in table 1. Mean (SD) age was 62.5 (8.3) years with mean (SD) diabetes duration of 10.3 (5.7) years. Most subjects had cardiovascular risks factors, including hypertension (80%), dyslipidaemia (76.9%) and obesity (73.3%). All subjects were previously treated with metformin while 58.5% were treated with BI.
Baseline demographic and clinical characteristics (mITT population, n=808)
Changes in HbA1c
The mean change in HbA1c at week 24 for the mITT population was −1.3% (95% CI: −1.4% to −1.2%), from 9.2% at baseline to 7.8% (p<0.001), meeting the assumption made in the hypothesis for this study of at least −0.4% decrease (figure 2A).

{kind=link}
{kind=link}
HbA1c ((A) overall, subgroups with previous OADs only and BI; (B) subgroups according to injection time) and FPG change ((C) overall, subgroups with previous OADs only and BI) from baseline to week 24. BI, basal insulin; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin; OADs, oral antidiabetic drugs.
Subjects previously treated with OADs presented a mean change in HbA1c at week 24 of −1.8% (95% CI: −2.0% to −1.6%), from 9.6% at baseline to 7.8%, whereas in the subpopulation previously treated with BI, the mean change was −1.0% (95% CI: −1.2% to −0.8%), from 8.8% at baseline to 7.9% (p<0.001 for both subgroups for baseline versus week 24 and p<0.001 for mean change between subgroups; figure 2A).
The subgroup of patients using iGlarLixi before breakfast had a numerically higher decrease in HbA1c between baseline and week 24 with a mean change of −1.5% (95% CI: −1.7% to −1.3%) compared with those using iGlarLixi before lunch, dinner or those who varied timing of administration during the trial, with a mean change of −1.3% (95% CI: −1.5% to −1.1%), −1.3% (95% CI: −1.5% to −1.1%) and −1.2% (95% CI: −1.4% to −0.9%), respectively (figure 2B).
The proportions of patients reaching HbA1c levels ≤6.5%, <7% and <7.5% after 24 weeks were 8.9%, 22.6% and 42.3%, respectively, in the overall population. Same proportions were 7.8%, 20.3% and 41.2% in the subgroup of patients previously treated with OADs alone, and 9.7%, 24.3% and 43.1% in those previously treated with BI.
For more than three-quarters of the participants, the individualised HbA1c% target levels were set to be lower than <7.5% (78.4%), For 14.7%, the target levels set by physicians were ≤6.5% at baseline.
The proportions of participants reaching at week 24 the individualised baseline-set target HbA1c in the mITT population were 36.1% (95% CI: 32.9% to 39.5%) in the overall population, 37.0% (95% CI: 32.8% to 41.4%) in the subgroup of patients previously treated with OADs and 35.5% (95% CI: 32.0% to 39.2%) in the subgroup of patients previously on BI.
Changes in FPG
From a baseline of 10.8 mmol/L, the mean change in FPG in the PP population (data available for 800 subjects) was −2.8 mmol/L (95% CI: −3.0 mmol/L to −2.5 mmol/L) at week 12 and −3.1 mmol/L (95% CI: −3.3 mmol/L to −2.8 mmol/L) at week 24 (p<0.001; figure 2C).
In the subpopulation of patients previously treated with OADs, the FPG decreased with a mean of −4.1 mmol/L (95% CI: −4.5 mmol/L to −3.7 mmol/L) at week 24, from 11.9 mmol/L to 7.8 mmol/L; whereas in the subpopulation previously treated with BI, the mean difference was −2.3 mmol/L (95% CI: −2.5 mmol/L to −2.1 mmol/L), from 10.0 mmol/L to 7.7 mmol/L at week 24, p<0.001 for week 24 versus baseline for both subgroups and p<0.001 for mean change between subgroups (figure 2C).
Changes in body weight
The mean body weight change for the total mITT population from baseline to week 24 was −1.6 kg (95% CI: −1.9 kg to −1.3 kg; p<0.001). A similar trend was observed for the subpopulations of patients treated at baseline with OADs (weight change −1.2 kg) and BI (weight change −1.9 kg) from baseline to week 24 (p<0.001 for both groups).
Changes in iGlarLixi doses and titration algorithms
At baseline, 76.9% of the PP population was prescribed iGlarLixi ratio 2/1 pen; and by week 24, this proportion decreased to 63.2%. An opposite trend occurred with iGlarLixi ratio 3/1 pen, with an increase from 23.1% to 36.8% of subjects recommended this pen.
For the iGlarLixi ratio 2/1 pen, the dose increased from a mean of 19.5 U/day to 30.2 U/day at week 24 (+10.7 U). In the case of the iGlarLixi ratio 3/1 pen, the dose increased from a mean of 30.1 U/day at baseline to 45.0 U/day at week 24 (+14.9 U).
The most prescribed titration algorithm for iGlarLixi in the PP population was algorithm B (60.6%), followed by algorithm A (16.6%) and algorithm C (7.4%). For 14.7% of patients, different titration algorithms were employed throughout the study. Algorithms C and A achieved the lowest mean value for HbA1c at week 24 follow-up, 7.6% (95% CI: 7.4% to 7.8%) and 7.7% (95% CI: 7.5% to 7.9%), respectively, but without a statistically significant difference compared with algorithm B (7.9%; 95% CI: 7.8% to 8.0%) or other algorithms (7.9%; 95% CI: 7.6% to 8.2%).
The titration was performed directly by the physician in 12.6% of study subjects at baseline and maintained in 10.6% at week 24. In most subjects (87.4% at baseline and 89.4% at week 24), self-titration was performed following physician instructions at baseline visit. The education of the subjects with T2D for iGlarLixi titration was done mostly face to face by the physician or nurse (87.8% at baseline and 85.2% at week 24).
Safety
Overall, treatment with iGlarLixi was well tolerated (table 2). A total of 43 (4.9%, 95% CI 3.7% to 6.6%) subjects reported 67 AEs.
Listing of adverse events (AEs) by systems organ class (SOC) and preferred term (PT)
Most frequent AEs were GI (nausea, vomiting, diarrhoea) reactions (2.1% of safety patient population) and hypoglycaemia (1.5%). No reactions at injection sites were reported. Only five SAEs were reported by four (0.5%) subjects.
Thirteen subjects (1.5%) reported a total of 14 hypoglycaemic episodes between baseline and week 24, corresponding to a rate of 0.04 events per PPY. Only one episode of severe hypoglycaemia was reported (0.1%), corresponding to a rate of 0.003 events per PPY. Detailed information regarding the occurrence of hypoglycaemic episodes is provided in table 3.
Hypoglycaemic events (safety population)
Discussion
In this large national non-interventional, prospective, RWE study, subjects with T2D suboptimally controlled with OADs±BI who initiated iGlarLixi achieved a significant change in the HbA1c values from baseline to week 24. Mean HbA1c decrease was −1.3% at week 24. As expected, HbA1c decrease was higher in the subgroup of patients previously treated with OADs only (−1.8%) compared with those previously treated with BI (−1.0%).
Despite the major differences in the design and conduct between RWE and RCTs, the glycaemic results of STAR.Ro are comparable with those in the LixiLan-O and LixiLan-L trials.19 20 In the LixiLan-O trial (including subjects with T2D not controlled with metformin with or without a second OAD), the mean change of HbA1c over 24 weeks of iGlarLixi treatment was −1.6% compared with −1.8% in STAR.Ro for the previously OAD treatment subgroup. It should be noted though that despite similar duration of T2D (8.9 years in LixiLan-O compared with 9.3 years in STAR.Ro), baseline HbA1c was higher in the STAR.Ro Study (9.6% for OAD subgroup) compared with LixiLan-O trial (HbA1c 8.1% at baseline).19 The higher magnitude of HbA1c decrease in the RCT is also supported by a subgroup analysis of results from the LixiLan-O trial,27 showing that in people with baseline HbA1c >9%, the mean HbA1c decrease was −2.9%, with 73.5% of subjects reaching HbA1c levels below 7%.
Similarly, change of HbA1c in the subgroup of patients previously treated with BI (−1.0% in the current study) was comparable with that recorded in the LixiLan-L trial (including patients treated with BI±OADs), which reported a mean HbA1c decrease of −1.1%.20 Again, despite similar T2D duration at iGlarLixi initiation (11 years in STAR.Ro vs 12 years in LixiLan-L), baseline HbA1c was higher in the current study (8.8%) compared with LixiLan-L (8.2%).
The higher baseline HbA1c recorded in STAR.Ro might partially explain the lower percentage of subjects reaching HbA1c target of <7%, especially for the OAD-only subgroup (20.3% for patients previously treated with OADs and 24.3% for patients previously treated with BI) compared with LixiLan trial subjects (74% for LixiLan-O, 55% for LixiLan-L).
The association between baseline HbA1c values and probability of reaching target HbA1c values with diabetes treatment was already firmly established.28 This might also explain why a higher percentage of subjects from the previously BI subgroup attained HbA1c targets in our study compared with subjects previously treated with OADs only.
In this observational study, the titration was mainly self-performed by the patients with healthcare practitioner oversight after in-person education session. Corresponding to real-life practice, no forced titration was followed over the study duration which is different with the protocol of the LixiLan RCTs. This led to a lower final dose of iGlarLixi for both 2/1 and 3/1 ratio pens (30.2 U/day for the 2/1 ratio pen and 45.0 U/day for the 3/1 ratio pen) in STAR.Ro compared with the LixiLan trials. Consequently, lower final FPG was obtained in the LixiLan trials (6.3 mmol/L for LixiLan-O and 6.8 mmol/L for LixiLan-L) compared with STAR.Ro results (final FPG of 7.8 mmol/L and 7.7 mmol/L, respectively). This might also explain the lower percentage of patients reaching HbA1c targets in STAR.Ro.
In STAR.Ro, iGlarLixi was preferably initiated with the ratio 2/1 pen (76.9%), pen that was maintained in 63.2% of patients at the end of the study (at the beginning of the study, only the pen with the 2/1 ratio was available in Romania, and the ratio 3/1 pen was available starting in June 2019). Mean body weight loss was considerably larger in the STAR.Ro (−1.2 kg for patients in the OAD subgroup and −1.9 kg in the BI subgroup) than in any of the LixiLan trials (mean change of −0.3 kg in the LixiLan-O and −0.7 in the LixiLan-L trial). It should be noted though that due to the observational nature of the trial, body weight was sometimes self-reported by study subjects so body weight change data should be interpreted with caution. On the other hand, means in STAR.Ro were not adjusted; whereas in the LixiLan trials, a mixed-effects model with repeated measures was used, providing estimates that are usually lower and closer to reality.
Frequency of hypoglycaemic episodes was remarkably low in the current RWE study. Thus, only 1.5% of subjects reported an episode of symptomatic or confirmed hypoglycaemia and only one patient (0.1%) reported an episode of severe hypoglycaemia. The rate of hypoglycaemic-documented symptomatic level 1 (<3.9 mmol/L) episodes was 0.02 events per PPY in subjects from the OAD-only subgroup of STAR.Ro compared with 1.4 events per PPY in LixiLan-O trial,19 and 0.009 events per PPY in subjects previously treated with BI from STAR.Ro compared with 3 events per PPY in the LixiLan-L trial.20 There can be several explanations, one being the observational nature of the RWE study, which makes it less precise in recording safety issues. In addition, lack of tight titration algorithm with final lower iGlarLixi dose and higher FPG levels in STAR.Ro might also account for this low frequency of hypoglycaemic episodes.
There are scarce published data reporting results of RWE studies performed in subjects treated with iGlarLixi, and these were usually performed in Southern and Eastern European countries. Thus, in the ENSURE Study performed in Italy,29 data on 675 patients with T2D initiated with iGlarLixi were reported. Baseline characteristics were slightly different compared with STAR.Ro data, with more males (54.2%) in the study group, having longer mean duration of diabetes (15.5 years) and being slightly better controlled with a mean HbA1c of 8.6%±1.4%. In the ENSURE Study, the mean reduction of HbA1c was −0.9% after 6 months comparable with that recorded in the STAR.Ro Study considering the lower HbA1c at treatment initiation. Similar reduction in weight (−1.21 kg) was recorded in the ENSURE Study.29 Similar to STAR.Ro results, frequency of hypoglycaemia was low, with no severe hypoglycaemic events recorded.
The iGL 6-M Study performed in Hungary included a lower number of patients (442, of which only 353 were included in the efficacy analyses) and a lower percentage of patients treated with BI+OADs at baseline (20.1% compared with 58.5% in STAR.Ro). Treatment duration was 6 months. Baseline HbA1c (8.9%±1.3%) and mean HbA1c decrease (−1.5%) were similar with those from STAR.Ro, presumably due to similarities in the population of patients and medical systems in the two considered Central/Eastern European countries. No severe hypoglycaemic episodes were reported in the iGL 6-M Study while the frequency of GI AEs was 1.4%, slightly lower compared with our data (2.1% in STAR.Ro Study). Weight loss was also recorded, with a mean decrease of −2.3 kg over 6 months of treatment.30
IDegLira is the other FRC of BI and GLP-1 RA available for the treatment of T2D, containing 100 U/mL of insulin degludec and 3.6 mg/mL of liraglutide. RCTs included in the DUAL Programme confirmed that IDegLira obtained better glucose control compared with its individual components, with low risk of hypoglycaemia and weight loss. No head-to-head RCTs are available for iGlarLixi and IDegLira.31 A number of RWE studies are also available for IDegLira. One of the largest observational cohort studies included 2432 participants from the Swedish national registry and reported a 1.0% mean decrease in HbA1c and a mean weight loss of −1.1 kg at 12 months after initiation of IDegLira. Subjects included in the Swedish study had similar age (61.3 years) but longer duration of diabetes (12.7 years) and slightly lower baseline HbA1c (8.9%) compared with those from STAR.Ro.32
The results of the STAR.Ro Study should be interpreted with caution since there are some limitations, in part inherent to an RWE design. In addition, selection bias may not be completely ruled out since inclusion of patients was absolutely at the sole decision of the prescribing physician. Moreover, the study did not include subjects previously treated with other GLP-1 RAs while SGLT-2i were discontinued at the time of iGlarLixi initiation, both indications being approved after the current study was designed and initiated. Thus, the results obtained might not be characteristic of the overall population of patients with T2D currently initiating iGlarLixi treatment in routine clinical practice. Self-reporting of hypoglycaemic episodes, without the use of paper or electronic hypoglycaemia logs or downloading of glucose metre data by investigators, might also explain the low frequency of hypoglycaemic episodes, especially compared with the rates reported in RCTs. Another limitation was that, despite not reaching FPG goals, the final iGlarLixi dose remained below the maximum dose of 40 U and 60 U for the ratio 2/1 pen and 3/1 pen, respectively, and possibly explaining the low rate of reported AEs, including hypoglycaemia rates. It should also be acknowledged that the 24-week study duration may be too short for full-titration in real-world practice.
The main strength of our study is the high number of subjects included, making STAR.Ro the largest RWE study of iGlarLixi reported to date, with the information provided being complementary to that offered by RCTs. In addition, to the best of our knowledge, it is the first study providing information on the glycaemic impact at the time of iGlarLixi injection. The subgroup of patients using iGlarLixi before breakfast had a numerically higher decrease in HbA1c between baseline and week 24 compared with those using iGlarLixi before lunch, dinner or those who varied timing of administration during the trial. According to an expert opinion, injecting iGlarLixi before breakfast may have some advantages: postprandial glucose excursions are typically larger after breakfast in most patients, lixisenatide administered with the morning injection can cover both breakfast and lunch (with the latter being usually the main meal in our population), and the risk of nocturnal hypoglycaemia may be lower.18 Nevertheless, the study was not specifically designed to test the difference according to injection timing and therefore this subgroup analysis is only generating a hypothesis that should be further tested in a specific RCT.
Conclusions
In conclusion, in a real-world setting, 24 weeks of iGlarLixi treatment provided a significant HbA1c reduction with a low hypoglycaemia risk and body weight loss in people with T2D inadequately controlled with OADs with or without BI, supporting the results previously reported by iGlarLixi RCTs.
Data availability statement
No data are available.
Ethics statements
Patient consent for publication
Ethics approval
The study was conducted in accordance with the ICH-GCP regulations and was approved by the National Bioethics Committee of Drugs and Medical Devices, Romanian Academy of Medical Science (reference number: 215 NP/14.01.2019). Data monitoring was provided by the sponsor. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors would like to thank Dr César García Rey (Optimapharm, Madrid, Spain) for providing medical writing support, and Sanofi Romania for sponsoring manuscript submission in accordance with Good Publication Practice guidelines.
References
Footnotes
CB, AC, B-MM, MM and CG contributed equally.
Contributors All authors made substantial contributions to the intellectual content of the paper, including the design of the study (CB, AC, B-MM, MM and CG), acquisition of data (CB, AC, B-MM, MM and CG), statistical analyses (CB and MM), interpretation of data (CB, AC, B-MM, MM and CG), and drafting and critical revision of the manuscript (CB, AC, B-MM, MM and CG). CB is the guarantor.
Funding The study and the preparation of this manuscript were supported by Sanofi Romania.
Competing interests CB declares honoraria from Sanofi, Novo Nordisk, Eli Lilly, AstraZeneca, Boehringer Ingelheim, Medochemie, Servier and Viatris; support for attending meetings from Servier and AstraZeneca; and participation on a Data Safety Monitoring Board or Advisory Board for AstraZeneca, Boehringer Ingelheim, Eli Lilly, Novo Nordisk and Sanofi. AC declares honoraria from Sanofi, Novo Nordisk, Eli Lilly, AstraZeneca, Boehringer Ingelheim, Medochemie, Servier, Gedeon and Medtronic; support for attending meetings from Boehringer Ingelheim, Sanofi and AstraZeneca; and participation on a Data Safety Monitoring Board or Advisory Board for Boehringer Ingelheim and Sanofi. B-MM declares honoraria from Sanofi, Novo Nordisk, Eli Lilly, AstraZeneca, Boehringer Ingelheim, Medochemie, Servier and Viatris; and support for attending meetings from Servier, Eli Lilly and Novo Nordisk. MM is an employee of Sanofi and may hold stock or stock options. CG declares honoraria from AstraZeneca, Boehringer Ingelheim, Eli Lilly, Medochemie, MSD, Novo Nordisk, Sanofi, Servier and Terapia; support for attending meetings from Boehringer Ingelheim and AstraZeneca; and participation on a Data Safety Monitoring Board or Advisory Board for AstraZeneca, Boehringer Ingelheim, Eli Lilly, Novo Nordisk and Sanofi.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.