Article Text

Abstract
Introduction Trauma causes 40% of child deaths in high-income countries, with haemorrhage being a leading contributor to death in this population. There is a growing recognition that fibrinogen and platelets play a major role in trauma-induced coagulopathy (TIC) but the exact physiological mechanisms are poorly understood.
Methods and analysis This is a prospective multicentre, open-label, randomised, two-arm parallel feasibility study conducted in the emergency departments, intensive care units and operating theatres of participating hospitals. Severely injured children, aged between 3 months and 18 years, presenting with traumatic haemorrhage requiring transfusion of blood products will be screened for inclusion.
Sixty-eight patients will be recruited and will be allocated to fibrinogen replacement using fibrinogen concentrate (FC) or cryoprecipitate in a 1:1 ratio. Fibrinogen replacement will be administered to patients with a FIBTEM A5 of ≤10. All other aspects of the currently used rotational thromboelastometry-guided treatment algorithm and damage-control approach to trauma remain the same in both groups.
The primary outcome is time to administration of fibrinogen replacement from time of identification of hypofibrinogenaemia. Clinical secondary outcomes and feasibility outcomes will also be analysed.
Ethics and dissemination This study has received ethical clearance from the Children’s Health Queensland Human Research Ethics Committee (HREC/17/QRCH/78). Equipment and consumables for sample testing have been provided to the study by Haemoview Diagnostics, Werfen Australia and Haemonetics Australia. FC has been provided by CSL Behring, Australia. The funding bodies and industry partners have had no input into the design of the study, and will not be involved in the preparation or submission of the manuscript for publication.
The use of viscoelastic haemostatic assays and early fibrinogen replacement has the potential to improve outcomes in paediatric trauma through earlier recognition of TIC. This in turn may reduce transfusion volumes and downstream complications and reduce the reliance on donor blood products such as cryoprecipitate.
The use of FC has implications for regional and remote centres who would not routinely have access to cryoprecipitate but could store FC easily. Access to early fibrinogen replacement in these centres could make a significant impact and assist in closing the gap in trauma care available to residents of these communities.
Outcomes of this study will be submitted for publication in peer-reviewed journals and submitted for presentation at national and international scientific fora.
Trial registration number NCT03508141.
- PAEDIATRICS
- TRAUMA MANAGEMENT
- Bleeding disorders & coagulopathies
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first study to compare the use of cryoprecipitate and fibrinogen in the paediatric trauma population.
Addresses an important aspect of trauma management that has the potential to improve outcomes through earlier recognition and correction of coagulopathy.
We investigate the use of fibrinogen concentrate which has the potential to benefit patients being cared for in regional and remote centres that do not have ready access to frozen blood products.
The proposed trial will lack power to find a statistically significant difference in the hospital mortality or blood product usage between groups, data from this study will inform future studies focused on these outcomes.
While the study is not statistically powered to detect difference in functional outcomes, feasibility of completing these assessments will guide future research with patient centred outcomes.
Introduction
Trauma remains a leading cause of death in children in high-income countries, with major haemorrhage significantly contributing to the mortality rate.1 In addition to the primary insult causing haemorrhage, trauma-induced coagulopathy (TIC) is recognised as playing a significant role in the mortality of patients with trauma, with growing evidence for the phenomenon in the paediatric population.2
The contribution of hypothermia, haemodilution and acidosis to traumatic coagulopathy have long been recognised. More recently, it has been noted that coagulopathy is present in severely injured patients in the absence of hypothermia and acidosis and prior to large volume resuscitation.3–5 Evidence for other mechanisms is emerging; most are part of a cascade of effects arising from endothelial disruption due to direct tissue injury, hypoperfusion and the effects of catecholamine surge.6 There is evidence that fibrinogen deficiency plays a significant role in TIC, particularly in children.7 Observational studies from the paediatric cardiac literature8 9 have also found that hypofibrinogenaemia correlates with bleeding risk.
Currently in Australia, cryoprecipitate is considered standard practice for fibrinogen replacement as part of a major haemorrhage protocol (MHP). Fibrinogen concentrate (FC) has many potential advantages over cryoprecipitate for fibrinogen replacement in the paediatric trauma context, particularly with regard to ease of access and administration facilitating more rapid therapy along with consistent FCs in the dose delivered. Despite its potential practical advantages including ease of storage and consistent dose calculations, there are some significant disadvantages to FC use when compared with cryoprecipitate. Most notably, FC does not deliver other haemostatic factors which are contained in cryoprecipitate including factor VIII,factor XIII, von Willebrand’s factor and fibronectin. The role and potential benefits of these additional factors remains unclear in paediatric traumatic haemorrhage.
Retrospective data in children have demonstrated higher levels of fibrinolysis in children when compared with adult patients.10 11 The pathophysiological basis for this difference remains unclear but may contribute to differences in patterns of haemorrhage and high rates of hypofibrinogenaemia at presentation in children with traumatic injuries.
Early replacement of fibrinogen in paediatric massive transfusion (>40 mL/kg) using cryoprecipitate within 4 hours of ED presentation has been reported to significantly reduce 24-hour mortality in a propensity weighted retrospective cohort study (adjusted difference −6.9%; 95% CI −10.6% to −3.2%).12 This effect is reported to extend to 7 days for children who receive >100 mL/kg of red blood cells (adjusted difference −7.7%; 95% CI −15.0% to −0.5%) and children with penetrating injuries (adjusted difference −9.2%; 95% CI −15.4% to −3.0%).12
The time taken to deliver fibrinogen replacement has been studied in the adult population, with a median time to delivery of 60 min for cryoprecipitate, with a significantly shorter time to delivery seen for FC (29 min).13 There are no published studies comparing FC and cryoprecipitate in the paediatric trauma population. Given the differences in mechanisms and patterns of injury, lower frequency of major traumatic haemorrhage in children, lower mortality, high rates of long-term morbidity, physiological differences, lower incidence of chronic comorbidities, lower prevalence of anticoagulant therapy and system-based differences in the management of paediatric trauma it is likely that adult data cannot be directly generalised to children.
In this protocol, we present a feasibility study for the early replacement of fibrinogen in paediatric traumatic haemorrhage to be conducted across a range of hospitals in Australia, including both major trauma centres and regional centres. The primary outcome of this study is time to fibrinogen replacement comparing cryoprecipitate and FC. In addition, we include outcomes which focus on the feasibility of the study design, participant recruitment and functional outcome data collection. Data from this feasibility study will be used to inform design of future large-scale clinical trials focused on patient-centred functional outcomes along and blood product usage.
Objectives
This study aims to:
Investigate the feasibility of early fibrinogen replacement in traumatic haemorrhage using either FC or cryoprecipitate in children.
Compare time to administration of fibrinogen replacement between FC and cryoprecipitate.
Investigate the effects of fibrinogen replacement (using either FC or cryoprecipitate) on fibrinogen levels during traumatic haemorrhage.
Methods
Study design and settings
This is a prospective multicentre, open-label, randomised, two-arm parallel study conducted in the emergency departments (EDs), intensive care units (ICUs) and operating theatres of participating hospitals. Hospitals participating include tertiary paediatric trauma centres, mixed tertiary trauma centres and regional EDs who care for children with major trauma. Hospitals with access to rotational thromboelastometry (ROTEM) testing for patients with trauma and have an MHP which includes ROTEM guided product replacement are eligible to participate.
Participants
Severely injured children, aged between 3 months and 18 years, presenting with traumatic haemorrhage requiring transfusion of blood products will be screened for inclusion. Inclusion and exclusion criteria are presented in table 1.
Inclusion and exclusion criteria
Randomisation can occur prior to receiving results of ROTEM testing. This pragmatic design has been adopted based on clinician feedback and aims to ensure that all potentially eligible patients are included in the study and based on prior experience that if randomisation is delayed, a higher proportion of patients are not enrolled. Due to this pragmatic design, some patients will be enrolled and randomised to the study but not require the study intervention. Details of how this will be accommodated in the analysis are contained in the ‘sample size and statistical analysis’ section.
Randomisation
Randomisation can be completed in the ED, ICU and OT of the participating hospitals. The patient will be screened using the inclusion/exclusion criteria. Once confirmed that the patient is eligible, they will be randomised using a password-protected, computer-based randomisation programme. The randomisation schedule will allocate patients in a 1:1 ratio with variable block sizes, stratified by site.
Study procedures and timeline
Following randomisation patients will be allocated to either fibrinogen replacement using FC or cryoprecipitate. All other aspects of the currently utilised major haemorrhage management at the local site remain unchanged for both groups.
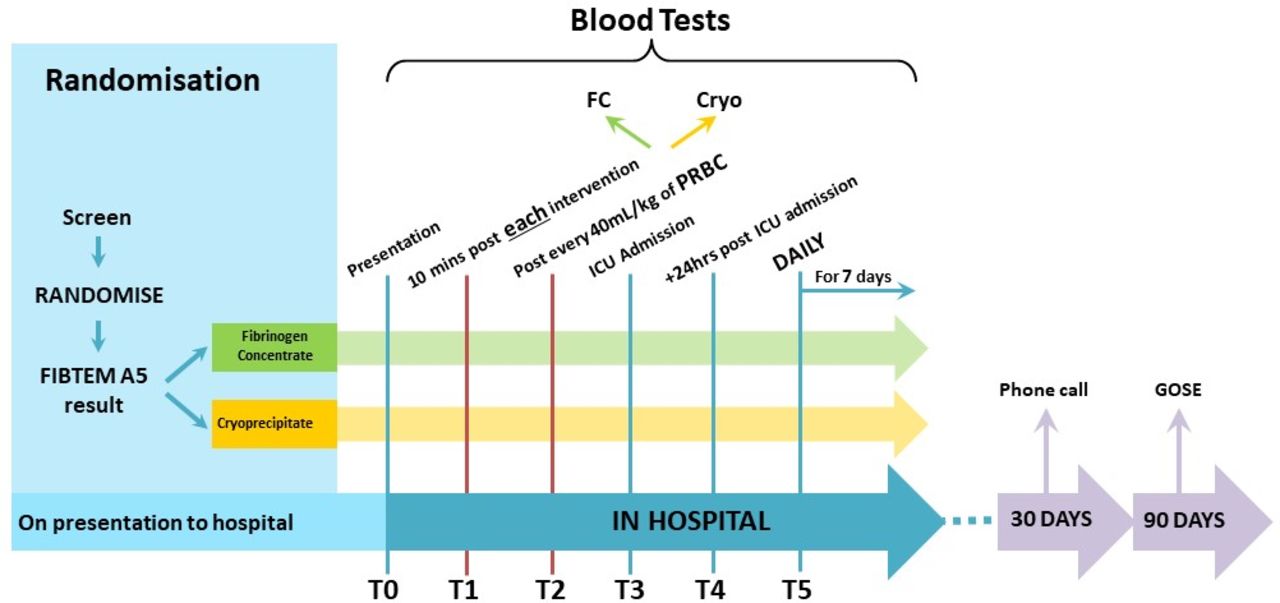
A sample for ROTEM testing will be collected and tested during the initial resuscitation on all children enrolled in the study. Fibrinogen replacement will be provided if a FIBTEM A5 result of less than 10 mm is returned (figure 1, table 2). ROTEM testing will be repeated 10 min after each episode of coagulation factor replacement, for each 40 mL/kg of red blood cells transfused or if there is a clinical concern prompting repeat testing (figure 1).

Study eligibility, flow chart and dosing schedule. MHP, major haemorrhage protocol; ROTEM, rotational thromboelastometry.
Dosing schedule for cryoprecipitate and fibrinogen concentrate based on FIBTEM A5
Children enrolled in the study will have repeat ROTEM testing on admission to the ICU, and then daily for 7 days or until ICU discharge, whichever is earlier (figure 2). Participants will be followed up at 30 days for survival and current location (in hospital, home, rehabilitation, long-term care) and at 90 days for functional outcome assessment using the Paediatric Extended Glasgow Outcomes Score.
{kind=link}
{kind=link}
Study timeline and intervention schedule. FC, fibrinogen concentrate; ICU, intensive care unit.
Treatment arms
Intervention arm
Fibrinogen replacement using FC as per ROTEM-guided treatment algorithm (FIBTEM A5 ≤10 mm), as per the dosing schedule (table 2).
Comparator arm
Fibrinogen replacement using cryoprecipitate as per ROTEM-guided treatment algorithm (FIBTEM A5 ≤10 mm) (table 2).
Study outcomes
Primary clinical outcome
Time to administration of first fibrinogen replacement from time of identification of hypofibrinogenaemia.
Secondary clinical outcomes
Blood product and coagulation factor usage at 6, 24 and 48 hours.
Duration of bleeding episode or time until surgical control and with no further requirement of blood or coagulation factors.
Duration of intensive care unit (ICU) and hospital length of stay (LOS).
Duration of mechanical ventilation.
Time to administration of fibrinogen replacement from time of arrival to trauma centre.
Effects of fibrinogen replacement on fibrinogen levels during traumatic haemorrhage as measured by FC and FIBTEM analysis.
Adverse events (AEs)—transfusion-associated circulatory overload/transfusion-related acute lung injury/multiorgan failure.
Thromboembolic complications.
All-cause mortality at 24 hours, 30 and 90 days.
Functional outcomes at 90 days.
Feasibility outcomes
Time to randomisation
Proportion of patients with blood sampling at all prespecified time points.
Number of missed patients (eligible but not enrolled).
Randomisation errors.
Protocol violations.
Follow-up rates with functional outcome assessment.
Data collection
Data will be collected using an assigned study number on paper case report forms by a trained research assistant. Where real time clinical data is required, a form will be provided to the bedside nurse for completion after training by the study team. Completed case report forms will be stored in a locked filing cabinet in the research office, accessible only to study personnel. A separate log of study numbers and patient identifiers will be kept in a separate locked draw in the research office, accessible only to study personnel. No identifying data will be entered into the database.
Study data will be entered into a web application specifically designed for the Fibrinogen Early In Severe paediatric Trauma studY (FEISTY) study by Research Path, it will have restricted access via authenticated login using an email address and password combination. Server hosting infrastructure for the web application maintains security requirements in accordance with international standards.
All sites will undergo regular on site and external monitoring to review compliance with the study protocol and ensure data accuracy and integrity.
As required by the Queensland State Archivist, all study information and documentation will be securely stored for a period of 15 years after the date of the child’s eighteenth birthday. As this trial will recruit participants down to birth age, all records will be securely stored for a total of 33 years before being securely destroyed.
Blood sampling volumes
Guidelines for the blood sampling in children for clinical research recommend a maximum of 3 mL/kg over a 24-hour period or 10% of total blood volume over a period of 8 weeks.14
Blood sampling is required for this study to allow for VHA analysis. All other bloods collected while on this study would form part of routine trauma care and are not considered to place an additional sampling burden to the participants in the study.
ROTEM testing is performed using one of two different analysers dependent on the sample volume collected. The minimum sample volume for the ROTEM delta is 1 mL and for ROTEM sigma is 3.8 mL. As such, where possible, the ROTEM delta will be used for all patients aged 4 years or less or where only small volume samples have been obtained.
The total blood volume collected over the first 24 hours of this study will be dependent on the severity of bleeding and product replacement. A maximum of 10 ROTEM tests will be performed in the first 24 hours for the purposes of this study, assuming the maximal sample volume of 3.8 mL for an adult size citrate tube this is 38 mL of blood. This equates to 3 mL/kg for a 12.5 kg child, correlating with a child of approximately 2 years of age. Given that adult size tubes are uncommon in this patient cohort it is more likely a paediatric tube would be used with a volume of 1 mL, thus a total of 10 mL over 24 hours, or 3 mL/kg of a 3.5 kg child (newborn).
After the first 24 hours, blood sampling for the purposes of the study are once daily for 7 days, thus falling well below the maximum of 10% of total blood volume over 8 weeks. All blood collection procedures will minimise discomfort for the child as much as practically possible.
Sample size
Data from the FEISTY adult trial has reported that the median time to administration for Cryoprecipitate is 60 min and for FC 29 min.13 While the time to administration in children may by slightly longer due to difficulty in IV insertion compared with adults, the difference between the two groups is expected to be similar. Assuming the use of survival analysis (log-rank test) for analysis of the primary outcome, a type I error of 0.05, power of 80%, equal sample sizes in the two study groups and an upper limit of 200 min for administration of either Cryoprecipitate or FC, 34 participants are required in each group (68 participants required in total). As previously described, some participants will be enrolled and randomised in the study and not require treatment with the study intervention. Thus, recruitment will continue until there are a minimum of 34 patients in each group who are eligible to receive the randomised study intervention as per the study flow chart (figure 1).
The proposed study and will lack power to find a statistically significant difference in the hospital mortality between control (cryoprecipitate) and intervention (FC) groups.
Statistical analysis plan
All analyses relating to the primary outcome and secondary outcomes will be performed on the modified intention-to-treat cohort comprising all patients who are consented, randomised and require treatment. Statistical analysis will be performed by a statistician blinded to the intervention allocation, prior to breaking of randomisation code. Descriptive statistics will be calculated for baseline characteristics, intervention characteristics and outcomes; mean (SD), median (IQR), number (percentage) will be presented dependent on variable type and distribution. No statistical comparisons will be undertaken to compare the baseline characteristics between the two study groups. The primary outcome (time to fibrinogen supplementation) will be compared between the FC and cryoprecipitate arms using Cox regression, including site as a random effect. The HR, 95% CI and p value will be reported. The primary outcome will also be reported for a per-protocol cohort, defined as those patients who are eligible as per the criteria above, consented, randomised and received their allocated treatment; only the HR and 95% CI will be reported. Additional time to event outcome measures will be analysed in the same way, without the reporting of a p value. Binary outcome measures will be analysed using logistic regression (allowing for site as a random effect) with OR and corresponding 95% CI reported; multinomial logistic regression will be implemented in a similar method for secondary outcome measures with more than two categories.
The differential trajectory of fibrinogen levels over time according to intervention will be assessed with fibrinogen measurements at predefined time points for each patient. First, exploratory analyses will be conducted using a ‘summary measures’ approach, comparing maximum and minimum fibrinogen levels within individual patients during the acute resuscitation phase and also the maximum fibrinogen in the post-resuscitation phase. Subsequently, bivariable and adjusted multivariable linear regression analyses will be used to assess the differential effects on circulating fibrinogen levels of treatment allocation using a generalised estimating equation approach with robust error estimates to account for the within-subject correlation of fibrinogen levels over time.
Analyses will be undertaken using Stata (StataCorp) with a significance level of 0.05. No adjustment for multiple comparisons will be made.
Data accuracy and integrity
All sites will undergo regular on site and remote monitoring. Monitoring will occur to define the study specific requirements and the agreed on commitments of the clinical study team involved in FEISTY Junior clinical trial operations. Monitoring visits will audit protocol adherence along with accuracy, completeness and the timeliness of data that is submitted to the study database and ensuring rights and safety of patients are protected and safety reporting is made to relevant authorities. Eligibility criteria will be audited for all enrolled patients and 10% of patients who are listed as excluded by the local site team. Source data verification will be undertaken for 100% of patients for the primary outcome, and a minimum of 10% of patients for secondary and feasibility outcome measures. Where data integrity issues are detected at a site, a full audit and source data verification will be undertaken for all patients at that site.
Patient and public involvement
Parents of children who have suffered from major traumatic injuries were consulted during the design of the project to determine the acceptability of the opt-out consent model. In addition, feedback on the acceptability of blood sampling and deidentified data collection were sought. All parents/guardians are given the opportunity to receive an email detailing the outcomes of the study at its conclusion during the opt-out consent discussion.
Ethical considerations
One of the primary challenges in performing research in an emergency setting is the inability to obtain true informed consent. Frequently, parents and guardians are not initially available when their child is brought into the ED. Furthermore, when parents or guardians are present, they are too distressed by the situation to comprehend study procedures and there is not enough time to obtain informed consent.
This study is presented with exactly these challenges:
Traumatic injury can represent a life-threatening emergency for which treatment is time critical.
There is often insufficient time to obtain informed consent within an appropriate time frame.
In the management of trauma patients staff priorities are assessment and management of airway, breathing, circulation with establishment of intravenous access and preparation of required equipment and medications.
Parents may not be present (eg, parents arriving separately to ambulance).
Even when parents and guardians are present, seeing their critically unwell child is very distressing and triggers considerable emotional distress, preventing the opportunity to present a research study in a calm, systematic manner.
Where possible, prospective informed consent will be obtained from the parent or guardian (online supplemental material 1). When prospective consent is not possible or practical, in keeping with the principles stated above, patients will be enrolled in the study using an opt-out approach. At an appropriate time after the stabilisation and resuscitation of the child, the parents will be approached by a member of the research team and presented with information regarding the study. They will have an opportunity to read the information sheet and any questions they have will be answered. At this time the parent/guardian will be provided with an opt-out form should they not wish for their child to continue in the study (online supplemental material 2).
Supplemental material
Supplemental material
Data collection for children whose parents or guardians choose not to continue in the study will be withdrawn from the study at that point, no further data will be collected, and treatment will return to standard practice at the local hospital.
This study has received ethical clearance from the Children’s Health Queensland Human Research Ethics Committee (HREC/17/QRCH/78).
Safety considerations
For the purposes of this study, the investigator is responsible for recording all AEs, regardless of their relationship to study drug, with the following exceptions:
Conditions that are present at screening and do not deteriorate will not be considered AEs.
Abnormal laboratory values will not be considered AEs unless deemed clinically significant by the investigator and documented as such.
The severity, expectedness and relationship of an AE will be assessed as per Common Terminology Criteria for Adverse Events V.4.0.15 AEs will be recorded for the duration of the acute admission to the trauma centre. Once the patient is discharged from acute care (ie, either home or rehabilitation) AE monitoring will cease. AEs will be documented from physical examination findings, clinically significant lab results or other documents that are relevant to patient safety. For patients randomised to the FC arm who experience AEs, these will be reported to HREC and CSL Behring as part of the contractual agreement between the FEISTY Junior study and CSL Behring.
Any serious AE (SAE) occurring in a study participant will be reported to the HREC within 24–72 hours of occurrence, in accordance with the safety reporting policy of the HREC. All suspected unexpected serious adverse reactions occurring in a study participant will be reported to the experimental drugs section, drug safety and evaluation branch of the TGA.
It is expected that all sites participating in the study will adhere to the highest standards of trauma care and prevention of deep vein thrombosis (DVT) or pulmonary embolism. This includes standard clinical surveillance of all major patients with trauma for venous thromboembolism. When there is clinical suspicion of DVT or PE, participating sites will further investigate patients to confirm diagnosis. Thrombotic events will be monitored for up to hospital discharge and reported separately in the eCRF. If the patient has received FC, then this will be reported to CSL.
Study oversight
The trial will be overseen by a trial steering committee (TSC), the membership of which will include: the chief investigators and at least two other investigators. The role of the TSC will be to monitor and supervise progress of the trial and review at regular intervals relevant information from other sources.
A data monitoring and safety board (DMSB) will be established prior to the commencement of the trial consisting of a statistician and two independent clinicians. The members of the DMSB will be appointed once the study has authorisation to commence. Prior to the commencement of the study the DSMB terms of reference and standard operating procedure will be developed and include an independent review of all SAEs. Blinded safety data will be reviewed by the DMSB after 20, 40 and 60 patients have been enrolled in the study. There will be an independent review of all deaths enrolled into the trial by the DMSB. All data will be deidentified.
The trial will be coordinated at each site by an on-site study management team consisting of, at a minimum, the site PI and a site study coordinator/research assistant (‘research assistant’). The site PI will be responsible for local oversight of the study including: monitoring safety; ensuring that the study is conducted according to the protocol and ensuring data integrity. The site PI will review the data for safety concerns and data trends at regular intervals, and will promptly report to the HREC and the central coordinating team any significant protocol deviation or any other significant event or problem that arises during the conduct of the study
Dissemination
Outcomes of this study will be submitted for publication in peer reviewed journals as well as submitted for presentation at national and international scientific fora. At the conclusion of the trial, deidentified datasets will be available from the corresponding author on reasonable request subject to local ethical and regulatory requirements. An analysis plan with full statistical code will be develop and published in an open access forum prior to database locking. Deidentified patient level data will be provided on reasonable request, provided they comply with legislative and ethical requirements.
Significance
The use of viscoelastic haemostatic assays (VHA) and early fibrinogen replacement has the potential to improve outcomes in paediatric trauma through earlier recognition of TIC. This in turn may reduce transfusion volumes and downstream complications and reducing the reliance on donor blood products such as cryoprecipitate.
The use of FC has implications for regional and remote centres who would not routinely have access to cryoprecipitate but could store FC easily. Access to early fibrinogen replacement in these centres could make a significant impact and assist in closing the gap in trauma care available to residents of regional and remote communities.
This is the first study to compare FC to cryoprecipitate in this population, important clinical data regarding time to replacement and the trajectory of fibrinogen levels in this population will better inform clinical practice. In addition, this feasibility data will inform the design of a larger clinical trial focused on patient-centred functional outcomes and blood product utilisation.
Current status of trial
This study commenced recruitment in July 2018. The current version of the study protocol is version 2.2, dated 21 July 2020. Recruitment to the required sample size is expected to be completed by December 2022.
Ethics statements
Patient consent for publication
Acknowledgments
The authors would like to thank the parents and children participating in this trial and the the medical, nursing and research teams in the participating sites for their help in study setup, recruitment, data collection and monitoring of study data.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @shanegeorge77, @lizrahilly1
Collaborators The FEISTY Investigators:Gold Coast University Hospital: Shane George, Elizabeth Wake, James Winearls, Don Campbell, Christa Bell, Megan King, Sarah Czuchwicki, Martin Wullschleger and the Trauma Service Case Managers. Queensland Children’s Hospital: Natalie Philips, Ann Hinde, Angus Jones, Tara Williams, Andrew Blanch, John Roy, Tona Gillen, Marlene Keen, Roy Kimble, Sharon Maconachie. Townsville University Hospital: Greg Wiseman, Leonie Jones. Mackay Base Hospital: Anni Paasilahti and the Trauma Service Case Managers. Cairns Base Hospital: Catherine Tacon. Rockhampton Hospital: Helen Miles. Princess Alexandra Hospital: Glenn Ryan, James Walsham, Chantelle Judge, Rob Eley. Royal Brisbane and Women’s Hospital: Catherine Hurn, Frances Williamson and the Trauma Service Case Managers. Royal Adelaide Hospital: Daniel Ellis, Daniel Harris, Stephanie O’Connor. Women’s and Children’s Hospital Adelaide: Subodh Ganu. Westmead Children’s Hospital: Melanie Jansen, S Soundappan.
Contributors SG, JW and EW conceived the study and developed the initial protocol, with feedback after critical review from all other authors. MJ, AP and GW provided expert intensive care advice to refine the protocol development. SM provided anaesthesia expertise and input to the protocol design. JR provided paediatric haematology expertise and knowledge to the protocol design. KG provided statistical input to the study design and development of the statistical analysis plan. All authors have had input into this manuscript and approved the final version. All listed authors meet ICMJE criteria for inclusion as an author of this manuscript.
Funding This research is supported by research funding from the Emergency Medicine Foundation (EMPJ-371R27-2017), Metro South Health Trauma and Disaster Management Project Grants (2019_George_TRADIM) and Gold Coast Hospital and Health Service Study, Education and Research Trust Account. Equipment and Consumables for sample testing have been provided to the study by Haemoview Diagnostics, Werfen Australia and Haemonetics Australia. Fibrinogen Concentrate has been provided by CSL Behring, Australia.
Disclaimer The funding bodies and industry partners have had no input into the design of the study, and will not be involved in the preparation or submission of the manuscript for publication.
Competing interests JW has received educational, travel and research support from TEM International and CSL Behring.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.