Article Text

Abstract
Introduction Patients with advanced cancers often face significant symptoms from their cancer and adverse effects from cancer-associated therapy. Patient-generated health data (PGHD) are routinely collected information about symptoms and activity levels that patients either directly report or passively record using devices such as wearable accelerometers. The objective of this study was to test the impact of an intervention integrating remote collection of PGHD with clinician and patient nudges to inform communication between patients with advanced cancer and their oncology team regarding symptom burden and functional status.
Methods and analysis This single-centre prospective randomised controlled trial randomises patients with metastatic gastrointestinal or lung cancers into one of three arms: (A) usual care, (B) an intervention that integrates PGHD (including weekly text-based symptom surveys and passively recorded step counts) into a dashboard delivered to oncology clinicians at each visit and (C) the same intervention as arm B but with an additional text-based active choice intervention to patients to encourage discussing their symptoms with their oncology team. The study will enrol approximately 125 participants. The coprimary outcomes are patient perceptions of their oncology team’s understanding of their symptoms and their functional status. Secondary outcomes are intervention utility and adherence.
Ethics and dissemination This study has been approved by the institutional review board at the University of Pennsylvania. Study results will be disseminated using methods that describe the results in ways that key stakeholders can best understand and implement.
Trial registration numbers NCT04616768 and 843 616.
- ONCOLOGY
- Protocols & guidelines
- HEALTH SERVICES ADMINISTRATION & MANAGEMENT
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
PROStep is one of the first randomised trials to assess the impact of clinician and patient nudges based on patient-generated health data (PGHD) on symptom and functional status understanding among patients with advanced cancer.
Our randomised study design assesses the utility of clinician-directed PGHD information display, with or without a patient-directed active choice question, on communication about symptom burden and functional decline.
Our design allows novel longitudinal evaluation of the association between objective step counts and downstream use and outcomes.
A key limitation is that relying on data from weekly symptom reports and wearable accelerometers may select for a population that is healthier and/or younger than the general population of patients with advanced cancer.
Introduction
Patients with advanced cancers often face significant symptoms from their cancer and adverse effects from cancer-associated therapy.1–3 In addition to suffering from high symptom burden, patients with incurable cancer experience declines in functional status and quality of life due to cancer progression or treatment.4–7 Adverse symptoms, quality of life and functional status are associated with lower survival, greater acute care use and higher financial burden for caregivers and the healthcare system.8–13 Identifying patients with high symptom burden, poor quality of life or poor functional status is thus critical to ensure high-quality care for patients with advanced cancer.14–16 However, in large prospective studies, oncology clinicians assess patient symptom burden only 40% of the time. Furthermore, in nearly three-quarters of cases where patient-reported and clinician-reported symptoms are not concordant, clinicians underestimate symptom severity.11 17–19
Patient-generated health data (PGHD) are routinely collected information about symptoms and activity levels that patients either report directly or passively record using devices such as wearable accelerometers.20–23 PGHD assessment may allow clinicians to identify patients with high symptom burden or poor functional status who would benefit from timely supportive care interventions.24 25 Patient-reported outcomes (PROs), health outcomes that are directly reported by a patient, are one example of PGHD.24–27 Routine PRO assessment in medical oncology can reliably improve symptom management, resulting in improvements in healthcare use, quality of life and even patient survival. As a result, PROs have been increasingly incorporated into routine oncology practice. PROs may be collected in the clinic on paper or via applications that link to the electronic medical record, with early trials suggesting high levels of adherence at 74.9% and 79.1% for weekly and monthly PRO questionnaires among oncology patients.28 29 However, limiting PRO collection to in-clinic visits in oncology may be too infrequent to comprehensively account for patients’ symptom burden. Technologies that enable remote PRO collection using questionnaires delivered via mobile phone applications may provide more granular and relevant information about symptom burden to clinicians.26 This is particularly timely during the COVID-19 pandemic, as remote symptom monitoring has grown, given the need to decrease face-to-face visits and subsequent exposure risk for patients with active cancer.30–32
In addition to quality of life and symptom burden, functional status is a critical element in determining a patient’s treatment and prognosis. Accurate measurement of functional status is challenging, as assessment via questionnaires usually differs between clinicians and patients.33 34 Passive activity monitoring via accelerometer-measured step counts may provide objective measures of functional status that can be trended over time to inform discussions about treatment and prognosis. Activity monitoring among patients with advanced cancer is feasible and associated with high levels of adherence in prior trials.35–38
While PGHD provides important clinical and prognostic data, a critical evidence gap is how to optimally integrate these data in clinical care to improve symptom management. Behavioural economic principles can inform optimal use of PGHD to improve symptom management.39 Clinician-targeted automated default email and text alerts about prognostic risk or evidence-based practice may improve guideline-based practice in oncology.40–42 Additionally, patient-targeted nudges using active choice—a behavioural economic method used to address delays in decision making by prompting an immediate decision between alternative choices—may lead to greater completion of high-value decisions such as cancer screening.43–45
No prospective trial has compared clinician and patient nudges informed by PGHD collection to better improve symptom management.40–42 The objective of this study was to test the impact of an intervention integrating remote collection of PGHD with clinician and patient nudges to inform symptom and functional status and communication understanding for patients with advanced cancer and their oncology team.
Methods and analysis
We describe the design and methods for a single-centre prospective randomised controlled trial to assess the impact of an intervention consisting of default information provision using dashboards containing PGHD to oncology clinicians (‘PROStep dashboards’), with or without a patient-directed active choice text message, on patient-reported and clinician-reported symptom understanding and communication among patients with advanced solid cancers.
Study hypotheses
The primary study hypothesis is that a clinician-targeted automated dashboard consisting of information about remotely collected PGHD (symptom burden, quality of life and functional status) will improve patient-perceived symptom understanding and communication compared with usual care. Secondary hypotheses are (1) the addition of a patient-directed active choice intervention based on their self-reported PGHD will improve patient-perceived symptom understanding and communication over and above PGHD dashboards to clinicians alone; (2) remote PGHD collection will be feasible and acceptable to patients and clinicians; and (3) passive activity monitoring will be feasible and acceptable to patients and clinicians.
Study setting
Recruitment for the trial is ongoing at the Perelman Center for Advanced Medicine (PCAM), a large tertiary academic practice at the University of Pennsylvania Health System in Philadelphia, Pennsylvania. We plan to recruit 125 patients who have incurable lung or gastrointestinal (GI) cancer and are undergoing systemic intravenous chemotherapy. Recruitment began in November 2020.
Eligibility criteria
The study is recruiting English-speaking patients at the PCAM who have a diagnosis of metastatic or stage IV GI or lung cancer. Patients must receive their oncology care at PCAM, be currently treated with intravenous chemotherapy (or planned receipt within 2 weeks) and have a capable smartphone (table 1). These criteria were selected with input from oncology clinicians who treat patients with GI and lung cancer to identify a population with incurable cancer who are undergoing intravenous chemotherapy and are the most likely to have uncontrolled symptoms or a poor prognosis.
Inclusion and exclusion criteria
The study cohort includes patients currently undergoing chemotherapy at PCAM. Patients seeking second opinions or undergoing their treatment at other sites are excluded as the text-based intervention cannot be delivered to other sites. Due to the intervention’s components (ie, PRO surveys, active choice text messages, utility surveys, Fitbit app, etc), we exclude non-English-speaking patients. We also exclude patients undergoing single-agent oral targeted therapy (eg, epidermal growth factor receptor antagonists) or single-agent checkpoint inhibitor monotherapy in order to enrich the study population for patients with high treatment-associated symptom burden. We exclude patients on active interventional trials—including an ongoing palliative care clinical trial among patients with thoracic malignancies—because such trials often already have a PRO collection mechanism that may confound the impact of the intervention. Finally, we exclude patients who are bedbound or wheelchair users because step data collected as a key feature of the intervention would not be expected to improve care for these patients.
Participant screening
On trial initiation, we obtained permission from GI and lung oncology clinicians to approach their patients regarding this trial. Each week, a trained clinical research coordinator (CRC) screens the electronic health records to identify eligible patients scheduled to see a GI or lung oncologist at PCAM in the following week. One study principal investigator (PI) (RBP) rescreens potentially eligible patients that the CRC identified to confirm eligibility. The CRC then approaches potential participants at their upcoming infusion visit.
Recruitment and retention
The CRC approaches patients who screen eligible during routine infusion visits at PCAM using a script to describe the purpose of the study, the randomisation process and interventions for each arm of the study. If unable to meet the patient in-clinic, the CRC can contact eligible patients via email with a similar script (see online supplemental appendices A,B). After eligibility is confirmed, the CRC uses an iPad to direct interested participants to an electronic portal to review and complete informed consent/Health Insurance Portability and Accountability Act of 1996 (HIPAA) authorisation form, enrolment form and a baseline PRO questionnaire form (see online supplemental appendix C). These are completed and stored on Way to Health (W2H), an automated information technology platform at the University of Pennsylvania that integrates wireless devices, conducts clinical trial randomisation and enrolment processes, delivers messaging (via text or email), delivers self-administered surveys and securely captures data for research purposes.46 W2H has been used in over 100 clinical trials inside and outside of the University of Pennsylvania.47–51 Each consenting patient completes an eight-question enrolment form that includes demographic characteristics (age, sex, race and education) and questions relevant to the trial (symptom management, activity level and comfort using text messages) (see online supplemental appendix D). The CRC asks eligible patients who decline consent to complete a limited consent form, providing authorisation to collect a questionnaire with an additional question (‘Why did you choose not to participate in the study?’) (see online supplemental appendices E and F). This nine-question survey allows us to explore if the study design may unintentionally exclude patients of older age, specific races, lower education, or decreased email or text message access or use. The CRC also assists with setting up W2H and their Fitbit device in person or by phone or a virtual meeting (a support partner is encouraged to attend).
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Randomisation and allocation concealment
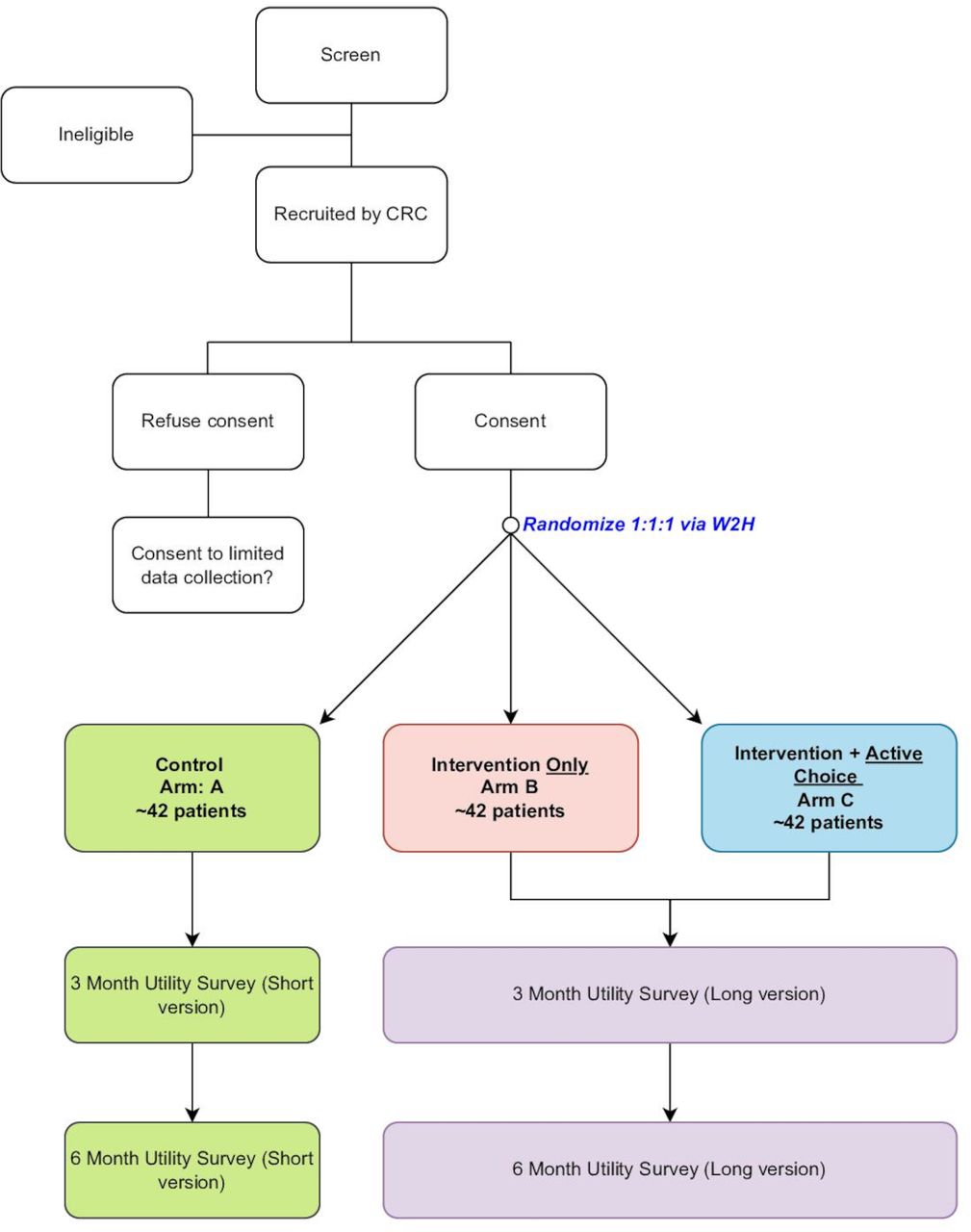
Patients are randomised in a 1:1:1 fashion into arms A, B or C (figure 1). Randomisation is completed electronically through W2H. After the patient enrols in the study and signs the consent, the patient completes the baseline demographic survey (as described previously) and W2H generates a link to the next step. If the patient is enrolled in arm A, the iPad includes no extra links and states that enrolment is complete. If the patient is selected for arms B or C, the participant’s W2H profile is sent an option to connect their Fitbit. The arms are randomly assigned by W2H using a random number generator, and arm assignment is given to the patient if asked. One of the study PIs (CRM) and the statistician (YZ) are blinded to patient randomisation.

Trial schema. W2H, Way to Health. CRC, Clinical Research Coordinator
Subject compensation
Patients in arms A, Band C are compensated with a $25 gift card for completing their first utility survey at 3 months after enrolment. Patients are eligible for a second payment of $25 (uploaded to their gift card) after completing their second and final utility survey at 6 months after enrolment. Patients in arms B and C are permitted to keep the Fitbit as part of the trial; Fitbits had a value of $80 on purchase. The CRC delivers a gift card to each participant shortly after they complete their 3-month utility survey and informs them to keep the card for the remainder of the study.
Interventions
The three arms in this trial include (A) usual care, (B) remote PGHD integrated into a PROStep dashboard delivered to oncology clinicians at each visit and (C) the same intervention as arm B but with an additional text-based active choice intervention to patients. Patients randomised into arm A will not have any intervention but will complete baseline and 3 and 6 month surveys. The trial does not dictate visit frequency, and patient encounter frequency across arms will be compared and reported in the main analysis.
PGHD collection
PROs
Once enrolled, patients in arms B and C receive a baseline PRO survey after they have consented and completed their enrolment questionnaire. These eight-question PRO surveys are sent to participants weekly on Monday mornings at 10:00 via text message on their mobile phone from the W2H platform. The text messages inquire about seven symptoms, which are selected from a list of 12 validated symptoms from the National Cancer Institute’s Common Terminology Criteria for Adverse Events and are scored on a 5-point scale from 0 (no present) to 4 (disabling); the final selection of symptoms was determined after focus groups with lung and GI oncology clinicians at PCAM.52 The patients also receive a validated question asking about their activity level over the prior month (table 2).53 For patients who do not respond to their PRO survey, automatic reminder alerts are sent on Tuesdays and Thursdays at 10:00 via W2H. If patients do not respond in 2 weeks, a CRC will follow up by phone. Weekly PRO data are reported in the dashboard delivered at each oncology visit and described as follows.
Pro Survey
Step count monitoring
Fitbits have been shown to accurately measure step counts in previous research studies.54–56 Each patient enrolled to arms B and C are given a Fitbit Inspire HR at enrolment. The CRC instructs patients on how to set up and wear their Fitbit and periodically sync the device with their phone to send step data to W2H. As the device has a 5-day memory, patients receive a reminder to synchronise their Fitbit two times per week as well as 2 days before a clinic visit unless the data have been synchronised in the prior 24 hours. If patients have no step data transmitted for a 2-week period, the CRC contacts the patient.
We collect step count data on a daily basis. As with weekly PROs, step count data are incorporated in the dashboards. Of note, GPS data are not collected nor used in this study.
PROStep dashboard
For patients enrolled in arms B and C, the patient’s medical oncology clinician—physician, physician assistant or nurse practitioner—will be given a PROStep dashboard in paper or electronic form at each visit after enrolment. These dashboards include (figure 2)
Summary report: brief text at the top of the dashboard that describes any severe symptoms (≥3) or abrupt worsening symptoms (change in ≥2 points from the previous survey) from the PRO surveys and/or a 10% decrease in step count from the previous week.
The date of the patient’s last outpatient palliative care visit, if any.
The date of the patient’s last documented serious illness conversation, a structured conversation about patient goals and end-of-life wishes, if any.57
PROs: table that displays patient responses to PRO questions over the last three surveys.
Step data: graph of average weekly step counts.
Acute care use: the number of oncology urgent care, emergency department visits and inpatient admissions in the past 90 days in the University of Pennsylvania Health System, as well as the date of the most recent event.
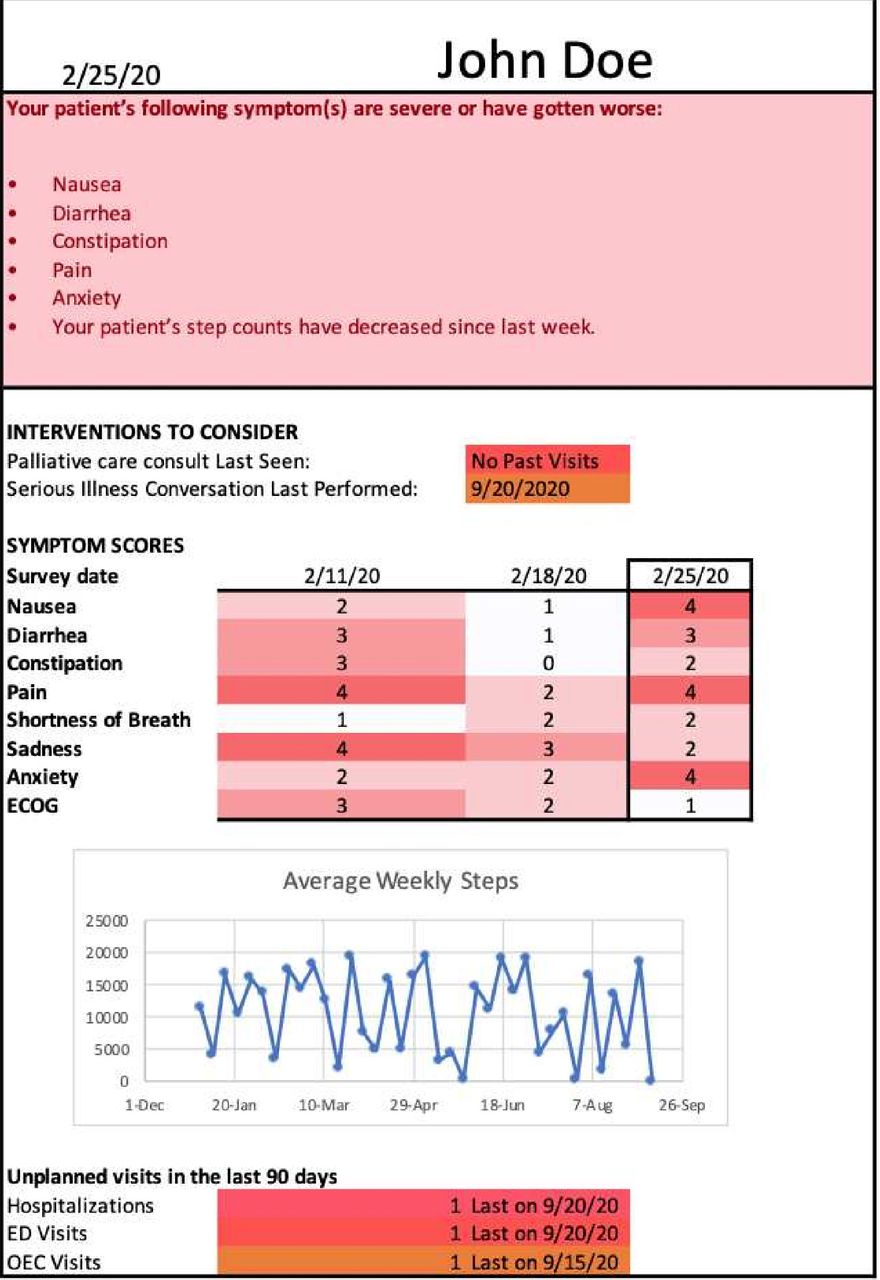
PROStep dashboard. ED, emergency department. OEC, Oncology Evaluation Center. ECOG, Eastern Cooperative Oncology Group performance status
To create these dashboards, W2H collects the weekly PRO responses and step counts sent by patients and automatically sends data for each patient to trained research personnel in the form of an Excel (version 2016) spreadsheet 1–2 days prior to their upcoming oncology appointment. Research personnel copy these data to an Excel template that generates the personalised dashboards and physically deliver a printout of each dashboard to clinicians’ offices on the morning of their appointment. These can also be sent electronically if staff is unable to physically travel to PCAM (as was often the case due to COVID-19 restrictions in place during some of the trial period).
Patient nudge to discuss symptoms
Patients enrolled in arm C receive an additional nudge prior to their oncology appointments. At 08:00 on the morning of every scheduled appointment, W2H automatically sends a text message summary of worsening symptoms based on their PRO surveys (ie, patient reports a severe symptom (≥3) or abrupt change in symptom severity (≥2)) and step data. This is followed by an active choice intervention consisting of the following question: ‘Do you plan on discussing these symptoms with your oncologist at your upcoming visit? type “1” if you plan to discuss them; type “2” if you do not plan to discuss them’. The purpose of this active choice intervention is to encourage patients to discuss their reported symptoms with their clinician at their upcoming appointment (figure 3).

{kind=link}
{kind=link}
{kind=link}
Intervention by patient arm. PRO, patient-reported outcome; W2H, Way to Health.
Data safety and monitoring
At the time of initiation of a new line of treatment, it is standard practice for patients with cancer to be given anticipatory guidance on when to seek medical attention. This practice will continue in this trial, and participants are reminded to contact their care team in the usual recommended fashion for any issues that arise during their care. They are also reminded weekly after each symptom report that they should contact their primary oncologist with any issues for which they think urgent medical attention is warranted.
Both the PI and CRC are notified if a participant reports a severe symptom (≥3) or any abrupt change in symptom severity (≥2), which also triggers an alert to the patient’s care team. In this way, multiple physicians will be aware of escalating symptoms.
Consent
On recruitment, the CRC contacts potential participants to confirm their eligibility and explain the study’s objectives, duration and requirements. Individuals who are interested in learning more about the study are directed to the W2H portal by the CRC, who uses an iPad to create a username for the new patient. On reaching the portal, potential participants are led through an automated online informed consent process (online supplemental appendix C). Successive screens explain the voluntary nature of the study, the risks and benefits of participation, alternatives to participation and the process for study withdrawal. On the final consent screen, a clearly delineated button enables patients to agree (or not) to participate in the study. Additionally, a platform electronic signature using a finger on a touch screen of a mobile phone will be required (online supplemental appendix D). Those who elect not to participate are asked to grant permission (or not) for the study team to complete a brief survey. An abbreviated study decline consent form is used for this purpose, similarly requiring the click of a clearly delineated button to agree (or not) to limited data collection and a platform electronic signature (online supplemental appendix E).
We received from the institutiona review board (IRB) a waiver of informed consent for consenting physicians. Prior to launching the study, the research team introduced clinicians to the trial at a weekly tumour board meeting for relevant GI oncology and thoracic medical oncologists. The PIs went over the study plan including the design, background and outcomes. We obtained verbal consent from all providers to (1) recruit patients into this study and (2) answer a utility survey at 3 and 6 months following enrolment for each patient. As the study does not limit clinical care in any way, clinician consent was obtained verbally at these meetings.
Outcomes
The two coprimary outcomes will compare responses between arm A and arms B+C for the following two questions asked of patients at 6 months after enrolment (or 3 months if the patient did not complete their 6-month survey) (table 3):
How well do you feel your oncology team understands your symptoms (eg, nausea, vomiting, weight loss, etc)?
How well do you feel your oncology team understands your activity level and ability to function?
Primary and secondary outcomes
These will be assessed on a 5-point Likert scale (1=not at all, 2=slightly, 3=moderately, 4=considerably, 5=completely). Of note, the prespecified comparison was between arm A and arm B and arm C, but the protocol was amended prior to any data review to combine the intervention arms due to slow enrolment and higher than expected dropout.
The secondary outcomes will compare these same two questions measured at 3 months between arm A and arms B+C. Another secondary outcome will be cumulative adherence between patients in the intervention arms at both 3 and 6 months. Patients are considered adherent for each week that they complete their PRO survey and sync their Fitbit step data for four or more days. Cumulative adherence is calculated when these weeks are divided by the total number of weeks that a patient was enrolled in the trial. We will also analyse trends in the PROStep data (ie, PRO survey scores and Fitbit step data) among intervention patients.
Exploratory outcomes will include multiple use metrics collected via the electronic medical record (EHR; i.e., number of ER visits, hospitalisations, palliative care consults, documented advanced care planning notes, and documented serious illness conversations. Patient utility will be another exploratory outcome measured using survey data at 3 and 6 months. Additionally, we will further analyse patient surveys by comparing the same two questions from the primary outcome between each individual arm and analyse responses to the remaining survey questions. Finally, we will measure clinician utility by comparing their survey responses at 3 and 6 months (or 3 months if the clinicians did not complete their 6-month survey for a specific patient).
Analysis plan
We will report descriptive statistics for patient characteristics in each arm, as well as their responses to the surveys of 3 and 6 months and PGHD.
For the two survey questions that compose the two coprimary outcomes, the study will compare the mean score from each survey question at 6 months across all three study arms using a Kruskal-Wallis test with p<0.05 indicating statistical significance. If the result is significant, the study will use Tukey’s honestly significantly difference test to test pairwise comparisons between the study arms. If the outcomes for any arm are skewed (not normally distributed), outcomes will be log-transformed before applying all tests.
For the secondary outcomes, a similar analysis will be conducted for the secondary outcome comparing the same questions but taken from the 3-month rather than the 6-month utility survey. We will apply t-tests to compare mean response scores for the remaining secondary outcomes. We will use Kruskal-Wallis tests for continuous outcomes and χ2 tests for categorical variables to compare the secondary outcomes of adherence rates and trends in PROStep data for arm B versus arm C at 3 and 6 months.
To assess whether responses in surveys of 3–6-months differ across arms, we will conduct an analysis of covariance model with the baseline score and arms as covariates, and change of score as the dependent variable.
Patients are welcome to discontinue the trial at any time, for any reason, via text, email, phone or at a visit with their oncologist. Patients who enrol in hospice will be disenrolled and will no longer receive surveys or prompts to reduce patient burden near the end of life, but we will use their most recent survey in the analyses of 3 and 6 months. If a patient exits the study early but more than 4 weeks after enrolment or 4 weeks after the 3 month survey, they will receive the utility survey of 3 or 6 months, respectively. This may not be possible for some patients who exit the study to enter hospice or due to death. If patients die or are otherwise unable to complete a survey, they will be omitted from the analysis for the relevant outcome. For patients who disenrol from the trial for any reason (voluntary, death, etc) but meet this 4-week threshold, their clinicians will receive a utility survey.
Statistical power
Power calculations were performed for the coprimary outcomes as originally specified prior to mid-trial protocol amendment: symptom and functional status understanding at 6 months between arms C versus A and arms B versus A. We estimated that there would be at least 80% power to detect a 0.68 increase on the Likert scale scores of symptom and functional status understanding. This estimate assumes that the baseline score was 3 (moderate symptom understanding) with a common SD of 1, and a significance level with a two-sided alpha of 0.025 for each coprimary outcome.
Patient and public involvement
Patient participants were not involved in the development of the research question or outcome measures. Patient and physician participants were involved in the design of the dashboards and text message wording. The utility surveys at 3 and 6 months include a short section of questions assessing the burden of the intervention among patients in arms B and C.
Discussion
Patients with advanced, incurable cancer who receive systemic therapy often have significant symptoms and declining functional status that is under-recognised and poorly managed by oncology clinicians. Remote electronic PRO and wearable step monitoring offer an opportunity to more accurately track and convey longitudinal information about symptoms and activity level for patients undergoing cancer treatment. This trial tests the impact of an intervention consisting of presenting remote PGHD via clinician dashboards, with or without a patient active choice intervention, on shared patient–clinician understanding of symptoms and functional status.
This study has several strengths, including (1) its patient-level randomised design; (2) enrolment of patients with two advanced cancers who often undergo chemotherapy and experience high symptom burden and functional decline during the course of treatment; (3) intervention that combines activity monitoring and symptom self-reporting to assess symptom burden and functional status decline; (4) use of patient and clinician behavioural prompts to improve communication about symptom management, rather than just passive measurement of symptoms; and (5) patient-centred primary outcomes that reflect patient’s perceptions of adequate clinician communication and management of symptoms and functional status.
Limitations of this pilot study include its (1) single-institution setting; (2) potential lack of generalisability to other cancers, particularly haematological malignancies; and (3) reliance on adherence to text-based PRO assessments and prompts. If successful, we plan on conducting larger, multi-institutional randomised controlled trials to test interventions consisting of PGHD collection and behavioural prompts. Additionally, real-world adherence to PRO collection is a limitation of many PRO collection programmes, and our incorporation of passive monitoring may offer an important mechanism of PGHD collection that is not dependent on a patient actively responding to a questionnaire. Thus, despite known limitations, this study is likely to provide novel data to guide the deployment of PGHD programmes in other outpatient oncology settings.
We hope that nudges to patients and clinicians based on longitudinal PGHD collection will make changes in patient symptoms, quality of life and functional status more transparent to oncology clinicians, leading to more informed discussions and decisions about the burdensome impacts of advanced cancer and its treatment. Future studies may extend this work to evaluate the role of PGHD in monitoring symptoms and functional status for populations excluded from this study, such as patients with other cancers, patients who receive non-chemotherapy anticancer drugs, patients who receive their chemotherapy remotely and patients who do not own a smart phone. Additionally, future studies should also assess caregiver perceptions towards this and other PGHD-related interventions, given the importance of caregivers in facilitating patient understanding of digital health tools and survey completion.
Ethics
This study has been approved by the University of Pennsylvania IRB (protocol #843616). This trial is registered with clinicaltrials.gov with the official title ‘A Feasibility Trial Using Remote Patient-reported Outcomes and Wearable Technology-reported Step Data to Compare Engagement, Utilisation, and Functional Status in Patients With Incurable Lung and Gastrointestinal Cancers’.
The potential risks to human subjects in this project include (1) risks of breach of confidentiality of personal health information, (2) risks of participants misinterpreting this tool as a means of quick communication with their care team and (3) risks of a breach of data for participating clinicians. To minimise these risks, our study employs numerous safeguards to protect human subjects. These include an experienced and well-trained study team, a robust informed consent process, state-of-the-art data security, and ongoing emphasis that the electronic symptom-reporting tool is investigational and not a replacement for usual means of communication with patients’ care teams.
Dissemination
We anticipate collection of data for all outcomes will be complete in December 2021. In addition to presentation at scientific meetings and publication in scholarly journals, we plan to leverage resources at Penn to place our results in the public domain where they can be openly discussed before any policy changes are recommended. This includes developing and implementing strategies to describe results in ways that key stakeholders can understand and implement.
Trial status
At the time of manuscript submission, 105 patients from the University of Pennsylvania clinics have consented to participate and have been randomised, and 82 are currently enrolled in the trial.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the University of Pennsylvania institutional review board (protocol #843616). Participants gave informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors RBP and CRM conceptualised the study design and obtained the funding. RBP, CRM, PEG, LMS, LNS and MSP contributed to the development of the study design. WF, JW, JW, NK, MK and MB were responsible for the development of the data collection platform, field testing of the study logistics, including clinic and subject recruitment. RBP, CRM, YZ and JC are responsible for the development of the analysis plan. WF, RBP and CRM drafted the first version of the manuscript. All authors read, edited and approved the final version.
Funding The authors are solely responsible for the design and conduct of this study, study analyses and the drafting of this paper. The funder, the Institute for Translational Medicine and Therapeutics, played no role in study design, collection, analysis and interpretation of data, writing of the report and the decision to submit the article for publication (grant/award number 400-4133-4-578849-xxxx-2446–4732).
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, conduct, reporting or dissemination plans of this research. Refer to the Methods and analysis section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.