Article Text

Abstract
Background Frequent whole blood donors have an increased risk of developing iron deficiency. Iron deficiency can have detrimental health effects when left untreated. Donation intervals are commonly too short to replenish iron stores and extending these reduces donor availability. Oral iron supplementation is known to shorten iron store recovery time but may also induce gastrointestinal complaints. We aim to optimise the effectiveness of iron supplements while minimising the risks of side effects. Therefore, we will evaluate the impact of different iron supplementation protocols in terms of dosage and frequency on ferritin and haemoglobin levels, gastrointestinal side effects, iron deficiency-related symptoms and donor return compared with placebo supplementation.
Methods Twelve hundred whole blood donors with ferritin levels ≤30 µg/L are included into a double-blind, randomised controlled trial. Participants are randomly allocated to one of six arms, administering capsules containing 0 mg, 30 mg or 60 mg of iron, either on alternate days or daily for 56 days. At baseline and 56, 122 and 182 days of follow-up, ferritin and haemoglobin levels are measured, and compliance, donor return, dietary iron intake, gastrointestinal, iron deficiency-related symptoms and general health are assessed by questionnaire.
Ethics and dissemination This study will provide a comprehensive overview of the effects of different frequencies and dosages of administration of iron supplements on iron status and health effects, thereby considering individual differences in treatment adherence and lifestyle. The outcome will provide scientific evidence to guide the debate if and how oral iron supplements may support the recovery of whole blood donors with low ferritin levels.
Trial registration number NL8590; The Dutch trial registry.
- blood bank & transfusion medicine
- anaemia
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is a large (n=1200), double-blind, randomised controlled trial to determine the optimal iron supplementation protocol in terms of both intake frequency and dosage.
Outcome variables include ferritin and haemoglobin levels, complete blood counts, iron deficiency-related symptoms and gastrointestinal side effects.
Participants are thoroughly characterised at baseline regarding social-economic status, medical background, physical fitness, dietary intake and smoking status.
A limitation is the limited number of follow-up visits; however, these time points are the most relevant, and temporal patterns are modelled based on earlier studies.
Introduction
Due to the haemoglobin (Hb)-bound iron loss during donation, regular whole blood donors are prone to developing iron deficiency, often in the absence of anemia.1 2 Iron deficiency without anaemia is characterised by reduced serum ferritin levels. It is highly prevalent among frequent whole blood donors, with 15.0% of the female and 9.4% of male Dutch donors having ferritin levels <15 µg/L.3 Reduced ferritin levels increase the risk of developing anaemia and are associated with iron deficiency-related symptoms, including fatigue, reduced exercise endurance, restless legs, pica (appetite for non-nutritious substances) and reduced neurocognitive functioning.4–8
At most international blood banks, including the Netherlands, Hb levels are measured before donation to safeguard donor health and blood product quality.9 However, Hb levels do not reflect the amount of stored iron, which is significantly impacted by whole blood donations.10 Therefore, ferritin measurements have become more common among blood banks to assess the donor’s iron storage. Sanquin-incorporated ferritin-guided donation intervals, deferring donors for 6 or 12 months when ferritin levels are ≥15 and ≤30 µg/L or <15 µg/L, respectively. These cut-off values are based on WHO standards, as described previously.3 However, donor deferral has been shown to demoralise donors and reduce donor return rates.3 11 12
Several studies have shown that oral iron supplementation reduces the postdonation recovery time of ferritin and Hb levels to predonation levels.13 14 While donors in the countries like the USA, Finland and Denmark are already advised about iron supplementation for iron storage recovery after donation, other blood services are hesitant, often due to ethical concerns.15–17 Kiss et al showed in a randomised controlled trial (RCT) that postdonation iron supplementation led to full recovery of ferritin levels within 56 days, whereas the non-iron supplementing group did not reach full recovery after 160 days.18
In several clinical trials, the iron supplement ferrous bisglycinate has been shown to result in a relatively high fractional iron uptake. This has been attributed to its iron-bound chelates that prevent the iron from binding to other dietary compounds (eg, tannins, catechols and phytates) that inhibit iron absorption.19–22 Furthermore, supplementation with ferrous bisglycinate has been shown to lower the risk of gastrointestinal discomfort compared with other iron formulations, possibly due to the increased uptake by intestinal mucosal cells, leading to less iron entering the colon.19 23 24 Therefore, ferrous bisglycinate is already used by the Danish blood bank for donors who suffer from gastrointestinal side-effects.25 26
Iron absorption in the duodenum and its entry into the plasma compartment is regulated by the peptide hormone hepcidin.27 It has been shown that iron supplementation leads to an increased hepatic production of hepcidin in iron-depleted women, causing a reduced intestinal iron uptake up to 24 hours postsupplementation.28 29 While the dosages of elemental iron used in previous studies range from 19 mg/day to 240 mg/day, lower dosed iron supplements are shown effective in recovering the iron stores postdonation.28 30 These findings suggest that alternate-day supplementation with low-dose ferrous bisglycinate capsules may lead to higher fractional iron uptake and fewer side effects compared with high-dose iron supplements taken daily or two times per day.
The effects of oral iron supplementation on iron deficiency-related symptoms in donors with low ferritin levels have currently only been studied in one non-blinded, non-randomised pilot study.31 While many studies have shown beneficial effects of iron supplementation on iron store recovery time after whole blood donation, the optimal intake frequency and dosages are still unknown.13 32–34 Similarly, the influence of dietary status and treatment adherence on postdonation iron store recovery using iron supplements have not been thoroughly assessed.
This study aims to determine the effect of iron supplementation on haemoglobin and ferritin levels, side effects, donor return and iron deficiency-related symptoms in whole blood donors with low ferritin levels, thereby comparing varying intake frequencies and iron dosages.
Methods and analysis
Setting
Sanquin Blood Bank is a non-profit organisation with a legal duty to collect, process and provide blood products throughout the Netherlands.35 Before every blood donation, the donor’s eligibility to donate is assessed using a donor health questionnaire.36 Donors should be in good health, aged between 18 and 79, and not at risk for any blood-borne infections. In accordance with European and international legislation, male and female whole blood donors with Hb ≤135 g/L and Hb ≤125 g/L (measured with the HemoCue 201, Angelholm, Sweden), respectively, are not eligible to donate for 3 months. Furthermore, routine ferritin measurements have been introduced since November 2017 for newly registered donors and at every fifth whole blood donation. Donors with ferritin levels ≥15 and ≤30 µg/L or <15 µg/L are deferred for 6 or 12 months, respectively.
Study population
Whole blood donors who have successfully donated before, donate at a participating blood bank location, are fluent in Dutch, and whose ferritin levels are measured during their next donation are invited by email to participate in the study. Donors are excluded from participation when regularly taking iron supplementation within the 3 months before enrolment. During their next donation, baseline measurements are taken, and a questionnaire is sent to donors who have indicated to be willing to participate. Inclusion into the trial is based on the baseline ferritin levels. Donors with baseline ferritin levels of ≤30 µg/L are eligible for the trial; for the other donors, the study ends after their next donation (ie, baseline visit). All exclusion criteria are presented in figure 1.

Flowchart of the donor inclusion for the baseline questionnaire and the randomised controlled trial.
Study design
This is a six-armed placebo-controlled double-blind, RCT. Of the donors included at baseline, only those with ferritin levels ≤30 µg/L are eligible for participation in the trial and are randomly allocated to one of the six trial arms. The arms vary in (1) intake frequency; high frequency, or daily supplementation, versus low frequency or alternate-day supplementation and (2) dosage; high dose (60 mg elemental iron) iron supplements, versus low dose (30 mg elemental iron) iron supplements versus placebo. The inclusion of donors is continued until each trial arm consists of 200 donors. We aim for an equal distribution of male and female participants (figure 2).

Schematic diagram of the participant randomisation. HD, high dose supplements (60 mg elemental iron); HF, high frequency supplementation (daily); LD, low dose supplements (30 mg elemental iron); LF, low frequency supplementation (alternate day); P, placebo supplements; RCT, randomised controlled trial.
Study procedures
Shortly before the baseline and follow-up visits, donors are asked to complete an online questionnaire through Castor 37 Electronic Data Capture (EDC), the Netherlands and are provided with a hyperlink to the Wageningen University & Research page for an iron-specific food frequency questionnaire (FFQ).
At baseline, donors will visit one of the selected donation centres for a regular whole blood donation. When donors meet all eligibility criteria to donate, blood samples are taken from the blood donation sampling pouch. The collected blood samples are sent to the Sanquin National Screening Laboratory in Amsterdam for analysis. At least two times per week, the donor database containing donor ferritin levels is examined, and participating donors with ferritin levels ≤30 µg/L are selected. These donors are randomised and will receive iron or placebo supplements and additional study information through postal mail. Donors with ferritin levels >30 µg/L are informed that their ferritin levels are sufficient and, therefore, they are not eligible to participate in the RCT. During the follow-up visits, blood samples are collected through venipuncture.
Intervention and timeline
For this study, ferrous bisglycinate iron supplements are used with capsules containing 0, 30 or 60 mg of elemental iron. Participants are asked to adhere to the study product supplementation protocol as strictly as possible for 56 days after the baseline visit. The supplementation protocol instructs the participants to take the capsules at least 3 hours before and after eating products or taking medicine that interfere with iron uptake (eg, dairy, coffee, tea, soy, antacids and antibiotics). Therefore, participants are advised to take the capsules shortly before going to bed and asked to refrain from taking any additional iron supplements.
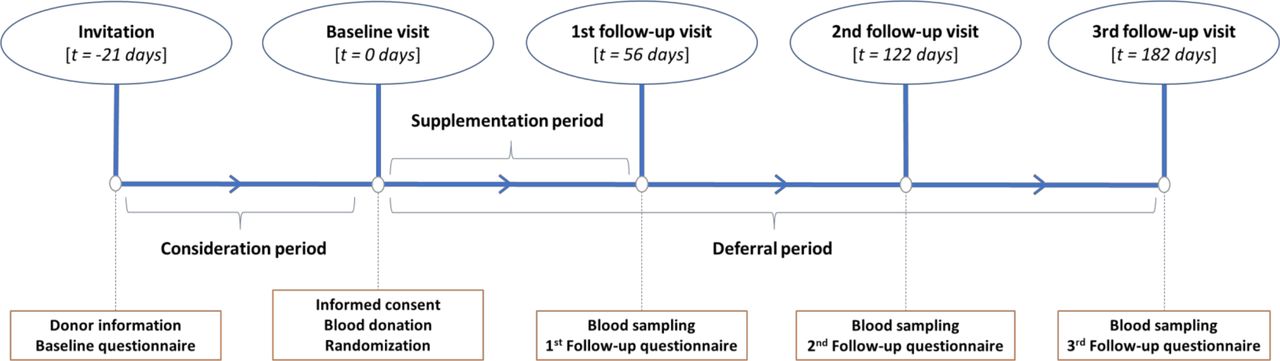
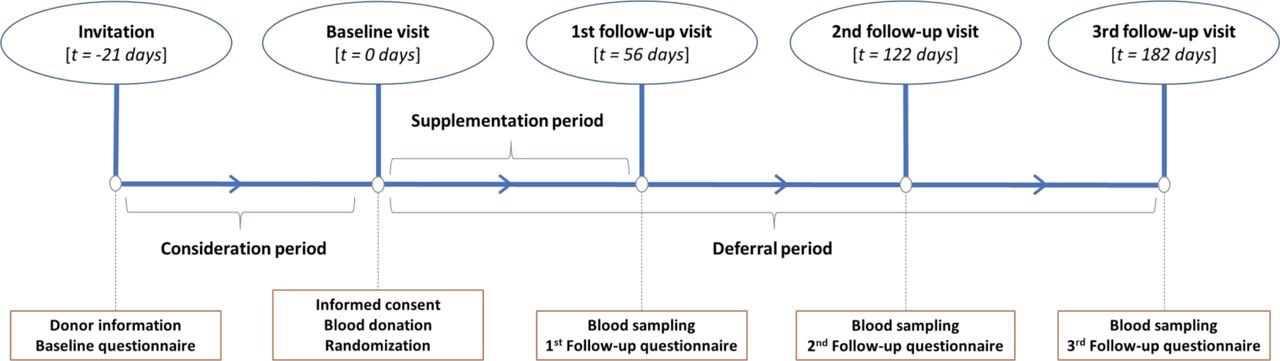
Follow-up visits will occur at 56, 122 and 182 days corresponding to the minimum donation interval for men, the minimum donation interval for women and the minimum ferritin-guided deferral period in the Netherlands, respectively (figure 3). The participants are asked to return any unused capsules at the first follow-up visit to determine their treatment adherence.
{kind=link}
{kind=link}
{kind=link}
Schematic diagram of the study timeline with t being the time since the baseline visit.
Sample size
Based on previous research performed by Kiss et al and Waldvogel et al, who used a similar randomised trial design in smaller groups of whole blood donors, sample size calculations were made for the primary outcome parameters, being ferritin and Hb, and the secondary outcome parameters, being adverse events, mental health and physical health (table 1).18 38 39 For the sample size calculation, we used the following formula:

Overview of the sample size calculations for Hb; ferritin; adverse events; mental health; physical health
Here, n1 is the sample size of the intervention groups, r is the ratio between the control groups and the intervention groups, σ2 is the variance of the continuous variable, v describes the expected difference between the continuous variables between the intervention and control groups (table 1),  is the two-tailed alternative hypothesis with significance level α=0.05 (1.96),
is the two-tailed alternative hypothesis with significance level α=0.05 (1.96),  is the probability of rejecting the null hypothesis when it is one minus probability of type II error (β) with a power of 0.90 (1.28).
is the probability of rejecting the null hypothesis when it is one minus probability of type II error (β) with a power of 0.90 (1.28).
It is expected that the chosen sample size is sufficient to reach statistical power for nearly all main outcome parameters (table 1).39 For mental health, a lack of power is expected. However, indications of trends towards an effect might be observed, and data may be used in future meta-analyses to reach statistical significance. Furthermore, we expect to observe small differences in outcomes between low-dose and high-dose iron supplementation groups. To observe an expected difference (v) of 4 µg/L in ferritin and 2.5 g/L in haemoglobin between the iron supplementation groups, at least 126 and 151 donors should be included in each intervention group, respectively. Therefore, to reduce the risk of underpowering the study and considering possible dropout, we will include 200 participants per intervention group. Moreover, this will allow us to perform subgroup analysis based on sex, treatment adherence and dietary intake.
Recruitment
Donors will first be recruited for the baseline questionnaire and blood sample measurements. The invitation is sent by email at least 3 weeks before the end of the donor’s standard donation interval or deferral period. Donors are provided with study information and an example of the informed consent form (online supplemental appendix A). The information folder contains information about the study procedures and the rights of the participant. The invited donors are asked to respond to the invitation through email to indicate if they would like to participate in the study before signing the informed consent form. During the blood donation visit (ie, baseline), donors who have agreed to participate are asked to sign the written informed consent form in a blood bank employee’s presence. By signing the informed consent form, donors officially agree to participate and confirm that their personal information and material can be used for research purposes. Finally, the consent form is signed by the blood bank employee to affirm being the study representative, after which the donor can continue with the regular whole blood donation. Participation in this study is voluntary, and donors will not receive any compensation besides travel expenses as part of regular Sanquin Blood Bank policies.
Supplemental material
We expect that 50% of the donors recruited for the baseline measurements have ferritin levels ≤30 µg/L and are eligible to participate in the trial.3 Recruitment will continue until 1200 donors have been included in the trial. Data on demographics such as sex, age, donation history and region are collected from the blood bank information system eProgesa (MAK systems, Paris, France). Recruitment will start in July 2022. We aim to include all participants within 1 year and expect to finalise all follow-up visits at the end of 2022.
The trial is initiated at one blood collection centre. Based on donor response rates and inclusion rates, additional centres are added. The additional centres are added based on their capacity and ability to cope with the additional study-related workload, accessibility, the number of regularly donating donors and the availability of direct transport to the National Screening Laboratory of Sanquin. Based on previous studies performed at Sanquin Research, we expect a response rate between 50% and 75%.40 41 The expected response rate corresponds with the inclusion of 1614 to 2421 eligible donors for the RCT per year, based on recruitment from two large Sanquin Blood Bank locations. In case of lower response rates, donors from other locations will also be included.
Blinding and randomisation
Participant randomisation is done on the individual level using a block randomisation method. This procedure is realised through Castor EDC, with randomised block sizes set at 12 and 18. Furthermore, the randomisation is stratified by age (18–49 years vs 50 years and older) and sex to account for menopausal effects in women, differences in ferritin levels between men and women, and the impact of ageing on iron absorption.42–44 Supplements and information are sent to the participants based on the number corresponding with the group they are allocated to after randomisation. The responsible researcher is blinded for which group number corresponds with which study product. The 30 mg iron, 60 mg iron and placebo capsules are identical in terms of appearance and weight to guarantee the blinding of the participants and involved researchers. The participants will not be blinded for the varying intake frequency.
Data collection
Data are collected at baseline and during three follow-up visits. The baseline questionnaire consists of a combination of previously used or validated questionnaires. Participants are characterised by social-economic status, medical background, physical fitness, menstrual status and smoking, as described in previous research.41 To determine the effects of iron supplementation, we will use the 36-Item Short-Form Health Survey; to determine baseline status and changes in general health,45 the International Physical Activity Questionnaire Short Form; to assess changes in levels of physical activity,46 the Fatigue Assessment Scale; to assess changes in self-reported fatigue,47 donation intention-specific Theory of Planned Behavior questions; to determine if donors are more or less willing to donate as a consequence of iron supplementation,48 and the Gastrointestinal Symptom Rating Scale and the Bristol Stool Chart; to assess the gastrointestinal side effects potentially caused by iron supplementation.49 To determine any effects of iron supplementation on iron deficiency-related symptoms, we use the Cambridge Hopkins Restless Legs Syndrome Questionnaire; to assess the presence of and changes in restless legs, the pica questionnaire; to assess a potential appetite for non-nutritive substances, and the Cognitive Failure Questionnaire; to determine changes in cognitive function.50–52
Questionnaires that will also determine possible confounding and effect modification are the Treatment Adherence Questionnaire53 to determine the participants’ compliance with the intake protocols and an iron-specific FFQ to determine their dietary iron, macronutrient and micronutrient intake.41 54 55 The FFQ allows us to assess whether or not iron supplements are more effective for donors who have low dietary (heme) iron intake and will only be completed at baseline and before the final follow-up visit. The follow-up visit questionnaires are similar to the baseline questionnaire, excluding the questions related to demographic characteristics and the addition of the adherence questionnaire for the first follow-up measurement. Furthermore, participants are asked to use the MedApp (MedApp Nederland B.V., Eindhoven, The Netherlands, https://medapp.nl) to assist with compliance with the supplementation protocol and to accurately assess treatment adherence. The MedApp will provide participants with daily or alternate daily notifications to remind them about their capsule intake and to indicate if the intake protocol was successfully followed.
At baseline and during follow-up, whole blood and serum samples are collected using 2 mL and 6 mL coated EDTA (VACUETTE, K3EDTA, Greiner Bio-one International GmbH, Austria) and 3.5 mL and 5 mL serum separating (VACUETTE, Serum gel, Greiner Bio-one International GmbH, Austria) tubes, respectively. Ferritin measurements (Architect Ci8200, Abbott Laboratories, Illinois), using serum samples, are performed routinely within 24 hours after the donation. The Architect Ci8200 is calibrated yearly for ferritin measurements by the manufacturer (Abbott Laboratories) and traceable to the first WHO Human Liver Ferritin International Standard (80/602). Furthermore, quality assurance assessments are performed daily by the laboratory staff, using low (20 µg/L), medium (150 µg/L) and high (400 µg/L) ferritin quality controls, provided by the manufacturer. When the daily quality assurance measurements do not meet the predefined acceptance criteria, the Architect Ci8200 will be recalibrated by the manufacturer (Abbott Laboratories). Quality management is in accordance with ISO 15189. Complete blood count measurements, including Hb and red blood cell parameters (Advia 2120, Siemens Medical Solutions Diagnostics, Breda, the Netherlands), are performed within 24 hours after the whole blood samples are taken. DNA is isolated using 400 µl buffy coat from EDTA whole blood samples (QIAsymphony DSP DNA Mini Kit, Qiagen GmbH, Hilden, Germany)56 and stored at −20°C, for later use. Additional processed and aliquoted plasma and serum samples are collected from the EDTA and serum separating tubes, respectively. The additional samples will be stored at −80°C and used for potential poststudy measurements of parameters, which might affect the iron hemeostatis (eg, inflammatory markers).
Statistical analysis
Descriptive statistics are presented for intervention and control groups and for men and women separately as means±SD for normally distributed data and median and IQR in case of a skewed distribution. For the analysis of the primary study parameters, the following multiple regression model is used if all assumptions for the model are fulfilled:

Here, Intercept represents the expected mean value when all explanatory variables have a value of 0, Dose1 the low dose iron capsules (30 mg), Dose2 the high dose iron capsules (60 mg), both dummy variables have 0 mg as a reference, frequency the every other day versus daily intake and e the error term or difference between observed and expected values. The assessed assumptions are the normality-distributed random error term and the assumption of the equality of variance. All the analyses will consist of two-sided tests, with a p value <0.05 considered statistically significant. Statistical data analyses are performed with SPSS (IBM SPSS Statistics V.23.0 or newer) and R (R Core Team, Windows).57
Potential effect modification by age, Body Mass Index (BMI), donation history, dietary intake (eg, heme and non-heme iron, dairy products, alcohol, tea, coffee and total energy) and menstrual status is examined. These potential effect modifiers have been selected due to their role in iron hemoestasis.58 59 We will test for effect modification by adding the variable of interest and an interaction term with the iron different iron dosages and intake frequency to the model. A variable is treated as an effect modifier when it has an interaction term with a p value <0.10 for more than half of the associations. When effect modification is observed, stratified results will be reported in addition to the overall results. The following potential confounders are examined; age, sex, BMI, donation history, menstrual status, smoking, season, compliance with the study products, social economic status, ethnicity, recent infections (including Sars-CoV-2) and baseline ferritin and Hb levels. A change of more than 10% in the regression coefficient is considered confounding, and variables are added to all the models.
Monitoring
Study monitoring is performed by the TAPAS Group, an independent monitoring bureau. The FORTE project has been labelled as a negligible risk study by the involved medical ethics committee based on a risk assessment. Study monitoring will consist of an initiation visit, two monitoring visits and a close-out visit.
Patient and public involvement
Donors, as well as non-donors, are involved in the FORTE research project through focus group interviews. The focus group interviews will consist of interactive group discussions involving frequent donors; donors who have donated at least five times, new donors; donors who have signed up for donation but have not yet donated and blood bank staff, including donor physicians. During the focus group interviews, the participants are asked to discuss their perceptions, opinions and attitude towards iron supplementation, current or potential alternative blood bank policies regarding iron management and any potential effects of these aspects on their willingness to donate. Based on the outcomes of the focus group interviews, we will design a questionnaire to quantify the findings from the focus group interviews. The questionnaire is developed based on the recurrent constructs and topics observed during the group discussions and distributed among a larger group of donors and new donors. The results from the focus group interviews and the questionnaire are used to determine the optimal approach and potential areas of concern for implementing iron supplementation as a blood bank policy, if shown effective based on the trial’s outcome.
Ethics and dissemination
Ethical considerations
This study is performed according to the Declaration of Helsinki and Good Clinical Practice guidelines. The Medical Research Ethics Committee of the Academic Medical Centre Amsterdam has approved the study protocol (trial ID NL8590). All participants are asked to provide their written informed consent and are informed that they can discontinue participation at any time. Data collection is compliant with the General Data Protection Regulation, and all data are pseudonymised to prevent the identification of study participants. To ensure the donors’ safety, invasive actions such as venipuncture are performed by trained blood bank employees, following the routine blood donation protocols of Sanquin Blood Bank, the Netherlands, whenever applicable.
Dissemination
Study results are published in peer-reviewed journals after evaluation of scientific relevance and quality by the involved researchers. Furthermore, the results are presented at (inter)national conferences, shared with study participants and communicated with donors and different Sanquin departments. Data that can lead to the identification of the participants will not be published.
Significance and outlook
This study’s outcomes will provide evidence to lead the debate if and how iron supplementation should be implemented to support iron repletion for whole blood donors with low ferritin levels. Blood donation and adequate blood availability are of great importance to guarantee patients’ health in need of blood transfusions. However, frequent blood donation increases the risk of iron deficiency in repeat whole blood donors. To ensure sufficient availability of blood and blood products and safeguard donor health, the donors’ iron status should be monitored and managed appropriately. However, international uniformity among blood services regarding policies to address the donors’ iron stores is lacking. Therefore, the FORTE study aims to provide new insights regarding the efficacy of iron supplementation for whole blood donors with low ferritin levels. We do this by comparing the effectiveness of high and low-dose iron capsules versus placebo for daily and alternate-day supplementation protocols in whole blood donors, thereby investigating laboratory and health-related outcomes. The thorough characterisation of participating donors will add to an enhanced understanding of the effectiveness of iron supplements under various circumstances.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JK, KvdH, MGS designed the study protocol. JK wrote the manuscript with input from KvdH, FAQ, MGS, DWS. VMJN, HLZ and HHW were consulted for medical and ethical input.
Funding Sanquin supported this research project: Product and Process Development Cellular Products Grant, project id: PPOC19-02/1-239.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.