Article Text

Abstract
Introduction Nearly one-quarter of patients discharged from the hospital with heart failure (HF) are readmitted within 30 days, placing a significant burden on patients, families and health systems. The objective of the ‘Using Mobile Integrated Health and Telehealth to support transitions of care among patients with Heart failure’ (MIGHTy-Heart) study is to compare the effectiveness of two postdischarge interventions on healthcare utilisation, patient-reported outcomes and healthcare quality among patients with HF.
Methods and analysis The MIGHTy-Heart study is a pragmatic comparative effectiveness trial comparing two interventions demonstrated to improve the hospital to home transition for patients with HF: mobile integrated health (MIH) and transitions of care coordinators (TOCC). The MIH intervention bundles home visits from a community paramedic (CP) with telehealth video visits by emergency medicine physicians to support the management of acute symptoms and postdischarge care coordination. The TOCC intervention consists of follow-up phone calls from a registered nurse within 48–72 hours of discharge to assess a patient’s clinical status, identify unmet clinical and social needs and reinforce patient education (eg, medication adherence and lifestyle changes). MIGHTy-Heart is enrolling and randomising (1:1) 2100 patients with HF who are discharged to home following a hospitalisation in two New York City (NY, USA) academic health systems. The coprimary study outcomes are all-cause 30-day hospital readmissions and quality of life measured with the Kansas City Cardiomyopathy Questionnaire 30 days after hospital discharge. The secondary endpoints are days at home, preventable emergency department visits, unplanned hospital admissions and patient-reported symptoms. Data sources for the study outcomes include patient surveys, electronic health records and claims submitted to Medicare and Medicaid.
Ethics and dissemination All participants provide written or verbal informed consent prior to randomisation in English, Spanish, French, Mandarin or Russian. Study findings are being disseminated to scientific audiences through peer-reviewed publications and presentations at national and international conferences. This study has been approved by: Biomedical Research Alliance of New York (BRANY #20-08-329-380), Weill Cornell Medicine Institutional Review Board (20-08022605) and Mt. Sinai Institutional Review Board (20-01901).
Trial registration number Clinicaltrials.gov, NCT04662541.
- heart failure
- telemedicine
- adult cardiology
- health policy
- protocols & guidelines
- quality in health care
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study will provide rigorous evidence of the comparative effectiveness of two evidence-based postdischarge interventions on outcomes relevant to patients with heart failure and health systems.
We will use the INSIGHT Clinical Data Network, a National Patient-Centered Clinical Research Network site, to facilitate clinical data sharing between health systems.
The study will provide generalisable study findings from a large urban, racially, ethnically and sociodemographically diverse population of patients with heart failure in the USA.
Regulations pertaining to community paramedicine vary widely state to state, which could limit generalisability of study findings because this study is being conducted only in New York.
As a pragmatic trial, a potential limitation is heterogeneity in the implementation of the two comparator arms across multiple sites within two health systems.
Introduction
Heart failure (HF) causes a substantial burden on both health systems and patients because of preventable hospital admissions. Patients with HF commonly report persistent symptoms within a week of discharge including shortness of breath, fatigue and oedema of the lower extremities.1 Among Medicare patients, the all-cause 30-day readmission rates for HF are 20%–25%,2–4 which is the highest for any medical condition in the USA. Readmissions are not only costly, but they expose patients to the ‘toxicities of hospitalization’,2 which include stress and inflammation, negative effects on cognition and mood, and a subsequent increase in vulnerability to stressors.5 In response to the penalties imposed by the Centers for Medicare and Medicaid Services (CMS) Hospital Readmissions Reduction Program,6 7 health systems have implemented educational programmes, peer-support interventions, and structured telephone support, but none of these interventions have consistently reduced HF readmission rates.8
Among discharged patients, early outpatient medical follow-up can reduce readmissions and improve symptom management.9 10 The American College of Cardiology has developed the ‘See You in Seven’ initiative, which emphasises the importance of outpatient follow-up within 7 days as a strategy to improve outcomes.11 Common barriers include low appointment availability and poor access to convenient transportation.9 12 Additionally, many primary care physicians lack the support and resources to address acute complaints around the clock for older, medically complex and socially vulnerable patients, and thus refer some patients to the emergency department (ED). Novel strategies to facilitate early follow-up and avoid preventable ED visits and hospitalisations are urgently needed.
Telehealth interventions13 are one strategy to reduce readmissions by reducing transportation barriers and improving patient–provider communication in the immediate posthospitalisation period.9 10 14 15 To date, randomised controlled trials (RCTs) of telehealth interventions among patients with HF have not consistently demonstrated improvement in clinical outcomes.16–19 One explanation is that, even when problems are identified, additional diagnostic processes (eg, lab testing) and therapeutic interventions (eg, diuretic administration) are not possible in a purely telehealth context. By incorporating the ability to remotely address acute complaints, mobile integrated health (MIH) brings together the strengths of telehealth with an in-person visit and can offer a novel strategy to improve early follow-up care.
The ‘Using Mobile Integrated Health and Telehealth to Support Transitions of Care among Heart Failure Patients’ (MIGHTy-Heart) trial is comparing two postdischarge interventions, a transition of care coordinator (TOCC) versus MIH. The TOCC intervention consists of follow-up phone calls from a registered nurse (RN) within 48–72 hours of discharge to assess a patient’s clinical status, identify unmet clinical and social needs and reinforce patient education (eg, medication adherence and lifestyle changes).8
MIH uses the skills and training of community paramedics collaboratively with on-demand telehealth supervision by physicians13 to remotely manage medically complex patients at high-risk for readmission. Community paramedicine addresses acute symptom exacerbations, educates patients and performs preventive measures in the home (eg, medication reconciliation and fall prevention).20–22 Licensed emergency medical technicians and paramedics complete additional training to become community paramedics including didactic courses, case-based simulation, in-home and clinic-based observations, all with a specific focus in geriatric medicine. During in-home MIH evaluations, the paramedics perform a standardised assessment, then video conference with a telehealth physician who directs additional diagnostics (eg, point-of-care lab testing), therapeutics (eg, administering intravenous diuretics) or care coordination (eg, referral to special providers).
Objectives
The objectives of the MIGHTy-Heart trial are to compare the effectiveness of two postdischarge interventions, MIH and TOCC, on healthcare utilisation (eg, repeat hospitalisations at 30 days, emergency room visits); patient-reported outcomes (eg, quality of life (QoL), symptoms); and healthcare quality (eg, days at home) among patients with HF. The overall hypothesis is that participants in the MIH intervention arm will have lower 30-day hospital readmission rates and improved QoL compared with those randomised to the TOCC intervention arm.
Methods and analysis
Study design and sites
We are conducting a multicentre, pragmatic RCT comparing MIH and TOCC interventions. The trial is taking place at two academic health systems in New York City (NYC): NewYork-Presbyterian (NYP) and Mount Sinai. We are recruiting from multiple hospitals within each health system across four boroughs of NYC.
Patients are recruited either in-person or virtually in English, Spanish, French, Mandarin or Russian. If translation services are needed, we use virtual translation services (audio or video) through the hospital-specific vendors in each health system. When in-person, patients sign informed consent, complete baseline data collection and are randomised prior to hospital discharge (figure 1). When virtual, the informed consent, baseline data collection and randomisation occur by phone within 48–72 hours of discharge. The participant is then contacted by respective staff in each treatment arm (eg, the MIH programme coordinator, or a RN coordinator in the TOCC arm) within 48–72 hours of discharge to facilitate delivery of the assigned intervention.

Overview of study design. ED, emergency department; MIH, mobile integrated health; TOCC, transition of care coordinator.
Interventions
Mobile integrated health
MIH involves monitoring patients after hospital discharge and providing home-based interventions through community paramedics to deliver targeted, patient-centred care to patients. MIH ‘bundles’ interventions to address symptom exacerbations and other causes of unplanned readmissions among patients with HF.22 Multidisciplinary experts, including emergency physicians, nurses, and community paramedics provide monitoring and follow-up to support patients transitioning from hospital to outpatient care.
Training of the community paramedics to deliver the MIH intervention includes didactic lectures specific to HF and case-based learning to simulate the evaluation and treatment of patients with HF. The comprehensive training for community paramedics was developed collaboratively between cardiologists and emergency medicine physicians.
The specific goals of MIH are to facilitate care transitions by treating acute symptoms in the home and by connecting patients with outpatient care teams and services in close collaboration with their primary HF cardiologists. Any time after hospital discharge, participants, family members and members of their care team (primary care doctors, cardiologists, care managers) can request a home visit by contacting the MIH care coordinator who triages the patient over the phone and activates a community paramedic visit as needed. Once activated, community paramedics are dispatched to the home and perform a standardised assessment, including a physical examination, vital signs, home safety evaluation and medication reconciliation.
During the MIH encounter, the medics use a standardised assessment that includes questions specific to signs and symptoms of HF. An emergency medicine physician is then contacted via telehealth using a tablet computer with a mobile cellular Wi-Fi hotspot. During the telehealth encounter, the emergency medicine physicians can access clinical notes, discharge summaries and medication lists via the institutional electronic health record (EHR). Adjustments to outpatient medications can be e-prescribed and follow-up appointments can be scheduled with the patient’s cardiologist or other specialists as needed. Additional diagnostic testing (eg, ECG, point-of-care labs) can be performed, and medications, (eg, intravenous diuretics) can be administered. As needed, cardiologists are available for consultation via Epic secure chat or video conference. At the end of the telehealth visit, encounter summaries are sent to the patient’s care team through the EHR, including their cardiologist, to ensure continuity of care and closed-loop communication. If patients are not connected with cardiology care, they are referred to one by a MIH care coordinator.
Overall, the MIH programme aims to provide additional support to the existing care team and augmenting services that might already be in place by home health aides or visiting nurses. MIH leverages the advanced capabilities of community paramedics and real-time oversight by physicians trained in the acute assessment of patients with HF. By providing rapid, in-home expert assessment initiated by the patient at the point of need and providing treatment as needed, MIH integrates the complementary strengths of two interventions: telehealth and community paramedicine.
Transitions of care coordinator
The TOCC arm consists of a follow-up phone call from a nurse within 48–72 hours of discharge. During the call, the nurse assesses the patient’s clinical status, identifies unmet clinical and social needs and reinforces patient education (eg, medication adherence and lifestyle changes). If the nurse identifies a clinical emergency, the patient’s physicians and care teams are notified, and if warranted, transport to the ED is arranged. The nurse may also connect patients to a care coordinator or social worker for non-emergent clinical and social needs. In addition, the nurse reminds the patient about any follow-up appointments scheduled at discharge and connects eligible patients to assistance with transportation programmes. After this phone call, the patient’s primary care team is responsible for addressing any clinical and social issues that are identified either during regularly scheduled office visits or patient-initiated phone calls. The majority of TOCC calls are documented in EPIC so the TOCC intervention can be quantified systematically across the two health systems.
Patient and public involvement statement
We have involved patients and stakeholders in every aspect of this study using a Stakeholder Advisory Board led by a Patient Engagement Officer (CG). The Stakeholder Advisory Board has diverse expertise spanning firsthand experience as a patient or caregiver, clinical nursing, care coordination, emergency medicine, community paramedicine and hospital operations/management (figure 2). The board meets bimonthly for the first year and quarterly for the second and third year to discuss issues pertaining to study implementation (recruitment, follow-up) and dissemination of information about the study and findings to public audiences.

Modified accelerator model for Stakeholder engagement. MIGHTy-HEART, Using Mobile Integrated Health and Telehealth to Support Transitions of Care among Heart Failure Patients.
Eligibility
The study population will include adults who meet the eligibility criteria specified in table 1.
Inclusion and exclusion criteria
Outcomes
The primary endpoints are all-cause hospital readmissions and change in QoL (table 2). The secondary endpoints are hospital-free days (days at home), preventable ED visits, unplanned hospital admissions from the ED, quality metrics and changes in other patient-reported outcomes. Outcomes are evaluated using data from EHRs, Medicare and Medicaid claims and patient surveys.
Schedule of baseline questionnaires, study outcomes and data sources
Study procedure
Screening and recruitment
First, enrolment coordinators review the list of eligible patients through the Epic EHR. Each site in the study is using Epic to ensure that the same algorithm for screening is applied at each site. To ensure consistent review of eligibility criteria across enrolment coordinators and sites, enrolment coordinators follow a detailed protocol to confirm eligibility and check for exclusion criteria. Second, they confirm that a patient has not previously been enrolled, approached or declined to participate using a database of all previously approached patients. Finally, enrolment coordinators confirm the patient is appropriate to approach and consent with the patient’s nurse or physician. For any cases that are ambiguous or could pose a safety concern to the emergency medicine technician (EMTs) who are conducting the MIH visits, the Emergency Medicine physicians (BD or KM) are contacted to review the patient’s chart in advance of enrolment.
In-person recruitment
Enrolment coordinators approach eligible patients during their hospitalisation and complete an enrolment form in REDCap. If the participant declines to participate, the enrolment coordinator documents the reason for declining: (1) not interested/comfortable in participating in research in general; (2) project would involve too much work/follow-up for participants; (3) concerns about COVID-19; (4) other. If the patient agrees to participate, the enrolment coordinator proceeds with the informed consent and baseline questionnaires. Only participants who complete the baseline questionnaires and sign consent are enrolled and randomised into the study.
Languages
Patients are recruited to the study if they speak or read in English, Spanish, French, Mandarin or Russian. All study materials, including the informed consents and patient-reported outcome surveys, are available in these languages. When patients need a translation service, we use LanguageLine Interpreters in both health systems to support the enrolment, intervention delivery and follow-up for the study.
Virtual recruitment
Eligible patients who cannot be approached during their hospitalisation due to infection control precautions, or who are discharged before the team can approach them, are recruited virtually. Within 48 hours of discharge, enrolment coordinators contact participants by telephone for recruitment and enrolment. During this call, they obtain verbal informed consent, and participants complete baseline surveys, either independently using a web link to the REDcap survey, or with the assistance of the enrolment coordinator who reads the questions and completes the responses for the participant. On study completion, participants are contacted to participate in an interview over the phone or using Zoom.
Randomisation
Participants are randomised 1:1 to receive either the MIH or the TOCC intervention immediately following enrolment. Within each health system, randomisation is performed using separate site-specific randomisation lists created in advance of the trial using a random number generator. For each new patient who is eligible and consented, a treatment arm is assigned sequentially from the randomisation list, in order of enrolment in the trial. Cross-over is defined as a case in which a participant is randomised to the TOCC group, but later referred to MIH by their cardiologist or care team.
Blinding
The enrolment coordinators, study statistician, principal investigators and coinvestigators are blinded to the treatment arm. The research coordinator, who is not directly enroling patients, conducts randomisation and alerts the appropriate clinical teams when a patient has been randomised to a treatment arm. Additionally, the study statisticians are blinded to the treatment arm assignment and see only a generic allocation (‘Intervention A’ and ‘Intervention B’). Only those directly involved in the provision of clinical care within the MIH and TOCC interventions are unblinded to the treatment arm.
Data collection
Key outcomes, instruments and data collection schedules are provided in table 2. Data are aggregated from multiple sources within a single secure computing environment, the WCM Datacore, for merging and analysis (figure 3).

Data sources across theUsing Mobile Integrated Health and Telehealth to Support Transitions of Care among Heart Failure Patients study. CMS, Centers for Medicare and Medicaid Services; CP, community paramedic; EHR, electronic health record; NYP, NewYork-Presbyterian.
Baseline data collection
After participant’s complete the informed consent, they complete a REDCap baseline survey that includes contact information, demographic information (age, gender, race, ethnicity, education financial status), access to technology (‘Do you have a computer in your home? If you have a smartphone, which type? Do you access the Internet?), health literacy, loneliness and self-care.
Healthcare utilisation and additional clinical information
The study database will include Medicare/Medicaid claims data and EHR data available through INSIGHT,23 a Clinical Research Network (CRN) affiliate of the National Patient-Centered Clinical Research Network.24 Through INSIGHT, EHR data from participating institutions across NYC (including NYP/WCM and Mount Sinai) are aggregated using common data elements. These data will be used to evaluate healthcare utilisation, cost and quality, and will also provide contextual clinical information (baseline medical history, medications and information relating to the index admission prior to enrolment in the trial) to inform all analyses.
Patient-reported outcome surveys
Participants will use REDCap, an encrypted, secure survey software, to self-report sociodemographic characteristics at baseline, along with multiple PRO surveys about global and HF-specific symptoms, functioning, and QoL at baseline, 30, 60 and 90 days. PRO surveys will include the Kansas City Cardiomyopathy Questionnaire (KCCQ), Patient-Reported Outcomes Measurement Information System 29 item survey (PROMIS-29) and the Self-Care of Heart Failure Index V.7.2 (SCHFI). All participants who do not complete the electronic surveys at the follow-up timepoints will complete the final follow-up surveys using paper-based forms.
Follow-up time points
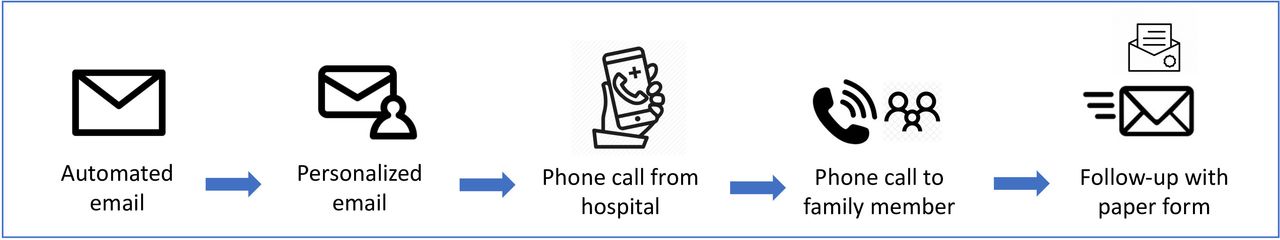
A study goal is to minimise missing PRO data through both the study design and statistical analysis. The PRO measurement system has been developed using design principles specifically intended to support sustained use among older adults. The study team will also provide additional support for patients with more advanced disease severity or trouble completing PROs. We have a multifactorial plan to support the completion of the follow-up study outcomes (figure 4), including:
REDCap automatically generates an email at 30 days follow-up.
RA (research assistant) sends personalised email after 32 days in case automated emails are filtered into spam because they are sent directly by REDcap.
RA calls the patient from a hospital phone number to help troubleshoot any problems they may be having and supports by completing the surveys over the phone in the language of their choice, using an interpreter service if needed. The RA follows up with three calls before calling a caregiver or family member, if they were included in the baseline survey as an alternative number.
There is an option for the RA to send participants hard copies of the questionnaires by mail for them to return in a self-addressed envelope.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Follow-up methods.
Data analysis
Missing data
Informative outcome missingness will be handled in the analyses by a doubly robust estimator (targeted minimum loss-based estimation (TMLE)), which uses two mathematical models: (1) the probability of having missing outcome data and (2) the probability of the outcome as a function of baseline variables. In this study, there are multiple risk factors for missing data: (e.g., disease severity, limited technology experience and death). The use of TMLE models corrects for differential missingness within strata of baseline variables that may be informative of the outcome. TMLE produces an estimator that is robust to missingness if at least one of these two models is correctly specified and is efficient if both models are correctly specified.
Coprimary outcome: all-cause hospital readmissions
The primary analysis will use time-to-event analysis methods to compare the all-cause hospitalisation rate within 30 days of hospital discharge between the two treatment arms (TOCC vs MIH). This will be determined by both Medicare Fee-for-Service claims and clinical data through INSIGHT CRN. Specifically, we will use TMLE25 to compute estimates of the hospitalisation rate at 30 days. TMLE uses an analysis method called covariate adjustment that leverages baseline patient characteristics to improve the efficiency of the effect estimates. Comparisons in the primary outcome between MIH and TOCC groups will be made using two-tailed tests based on the asymptotic Gaussian distribution of the estimators (at the 0.05 level of significance).
We will explore differences by site by performing stratified analyses to account for possible heterogeneity in the implementation of the intervention. Analysis for other prespecified subgroups of interest will be conducted for age, race, ethnicity, gender and healthcare utilisation (defined as the number of hospitalizations within the 12 months prior to study enrolment), obtained from Medicare and Medicaid claims data. Each subgroup analysis will be conducted separately comparing the two interventions. Rather than generate p values for treatment effects within each subgroup, the risk ratio (RR) point estimate and 95% CI will be generated for the treatment effect within each subgroup. Forest plots will be used to display the RRs and corresponding CIs. These will be examined for clinically meaningful differences across the subgroups to identify possible treatment effect heterogeneity. Missing data are expected to be minimal because the outcomes are measured with claims data.
Co-primary outcome: changes in QoL
All PRO-survey items will be scored and quantitatively assessed using standard descriptive statistics of frequency, central tendency and dispersion to describe the sample overall and by treatment arm. The primary analysis will be based on a comparison of KCCQ score means (SD) in the two treatment arms. We will use TMLE to compute the difference in means and to test for significant differences. Based on our prior work showing differences in these PRO surveys by patient characteristics, including age, comorbid conditions, and measures of psychosocial adversity, we will examine whether these patient characteristics are treatment effect modifiers.
Secondary outcomes: days at home
The primary quality outcome of days at home will be determined by both Medicare claims and clinical data through INSIGHT CRN. Days at home are emerging as an important quality metric from the patient perspective, as many patients want to maximise the number of days they can be home.26 This metric has also been associated with other important quality indicators such as use of hospitals and EDs, patient experiences and clinician morale.26 The primary analysis will compare the number of days at home within the 30 days following hospital discharge between the MIH and TOCC group. Comparisons in the primary outcome between groups will be made using TMLE (at the 0.05 level of significance). Forest plots will be used to display the estimated treatment effect across the subgroups and will be examined to determine treatment heterogeneity by identifying subgroups with clinically meaningful differences, such as HF severity.
Secondary outcomes: HF hospitalisations, preventable ED visits and unplanned hospital readmissions
We will use the same statistical approaches as described for the primary endpoints to also evaluate differences in unplanned readmissions within 30 days and preventable ED visits within 30 days. We will evaluate differences in unplanned readmissions within 30 days, using the 30-day all-cause unplanned hospital readmission algorithm from CMS, based on the patient’s index admission.27 This algorithm is used to calculate hospital performance in the Hospital Readmission Reduction Program and other Medicare pay-for-performance programmes. We will first identify all readmissions that occur within 30 days of the date of discharge for the index admission, and then exclude planned readmissions that are considered necessary for a patient’s care and clearly defined in this algorithm. All other readmissions will be categorised as unplanned. We will also evaluate preventable ED visits within 30 days, defined using the Billings/New York University ED utilisation algorithm between groups.28
Secondary outcomes: changes in other PROs
All PRO-survey items will be scored and quantitatively assessed using standard descriptive statistics of frequency, central tendency and dispersion to describe the sample overall and by treatment arm. Changes in symptoms will be measured using the PROMIS-2929 and SCHFI.30
Sample size considerations and power analysis
The target sample size in the parent trial is 2100. Power was computed using simulation methods. Because estimation in readmission rates generally required a larger sample size than estimation of changes in KCCQ scores, we first estimated the sample size required to detect a reduction of 5% (additive scale) in readmission rates at 5% confidence with 80% power. We then computed the power that would result from that sample size for detecting changes in KCCQ scores. Our sample size estimation procedure is based on a simulation using real data from the INSIGHT CRN. The simulation proceeds as follows. First, we identify all patients in our target population in the two healthcare systems, where the criteria for the target population are given in table 1. We then used this population to simulate a randomised study where a sample of size n patients is randomly divided into treatment and control groups. We then simulated a treatment effect of 5% by replacing the outcomes for a randomly chosen subsample of the treated population by a Bernoulli random draw, where the Bernoulli probability was chosen to achieve the desired 5% effect. In addition, we randomly dropped 10% of the outcomes to account for estimated attrition. In this simulated randomised study, we then computed the TMLE25, adjusting for sex, race, myocardial infarction, chronic kidney disease, chronic obstructive pulmonary disease, diabetes, pneumonia, respiratory failure and septicaemia. We repeated this procedure 1000 times and approximated the power as the proportion of simulated trials that correctly rejected the null hypothesis of no treatment effect. We repeated this procedure for sample sizes 1600, 1700, 1800, 1900, 2000 and 2100. We found that a sample size of 2100 would yield a power of approximately 85% for the effect of the intervention on 30-day readmission rates. This sample size will have 85% power to detect a change of 0.13 SD in global and HF-specific symptoms and QoL, measured with the KCCQ. The calculation for KCCQ score uses a two-group t-test with a 5% 2-sided significance level, and thus provides a conservative estimate of power compared with the TMLE methodology that will be used in the analysis of the data.31 This sample size yields sufficient power to detect meaningful differences in changes in KCCQ scores, that typically are considered as a 0.50 SD change, from baseline to 4 weeks, given that a 3–5 point difference in KCCQ is clinically meaningful.32
Ethics and dissemination
This study has been approved by the Biomedical Research Alliance of New York (BRANY #20-08-329-380), Weill Cornell Medicine Institutional Review Board (20-08022605), and Mt. Sinai Institutional Review Board (20-01901). All participants provide written or verbal informed consent prior to randomisation in English, Spanish, French, Mandarin or Russian. Study findings are being disseminated to scientific audiences through peer reviewed publications and are being presented at national and international conferences. The Stakeholder Advisory Board is providing guidance on the content and platforms for dissemination to public audiences.
Discussion
MIH has grown out of a need to better manage patients with HF in the transition from the hospital to home using optimised care coordination and real-time support when symptoms worsen. This need has been acknowledged nationally, with CMS recent payment model changes signalling a shift towards an expanded role for emergency medical services-based interventions.33 Nonetheless, there is an outstanding need to rigorously evaluate the effectiveness of MIH in a randomized controlled trial of patients with HF on multiple clinical and patient-centred outcomes. The results of this randomized controlled trial will provide rigorous evidence for patients, caregivers, clinicians and other stakeholders on the comparative benefits and risks of MIH and TOCC on outcomes that are meaningful to patients with HF.
Limitations
A limitation of this study is that there is the potential for study crossover, specifically that participants could be randomised to TOCC and then their physician refers them to the MIH group. This is likely to be a rare crossover event, but it is possible. For the MIH intervention patients have the choice to initiate a CP visit—all patients in the MIH arm do get calls from the MIH care coordinator and access to triage at the point of need. Additionally, as a pragmatic trial, there is heterogeneity in the TOCC arm by health system and physician practices within specific hospitals. We will conduct an intention to treat analysis based on randomisation to treatment arms, but we will also do per-protocol analysis based on the services delivered.
Ethics statements
Patient consent for publication
Acknowledgments
We would like to acknowledge the Patient Stakeholder Board who has provided input into the design and implementation of the MIGHTy-Heart study. We would also like to acknowledge the participants who are willingly taking part in this research.
References
Footnotes
Twitter @RuthMCreber, @ParagGoyalMD, @DhruvKhullar, @dslotwin
Contributors RMMC: contact study PI, codesigned the study including main conceptual ideas, drafted the manuscript. BD: study site-PI, codesigned the study, conducted pilot study. KM: study site-PI, codesigned the study. MRT: codesigned the study including main conceptual ideas, designed the figures. LST: implementing the study. CG: contributed to the study as a patient and caregiver stakeholder. ID: supported study design and proposal submission, will analyse the study data. PG: supported study design and proposal submission and pilot study. MW: leading data management team, designed study data infrastructure. JY, DK: supported study design and proposal submission. DS, KR: study site-PI. RK: study PI, codesigned the study including main conceptual ideas, is coleading the study.
Funding This work was supported by the Patient-Centred Outcomes Research Institute (PCORI), Award HS-2019C2-17373. 1828 L L Street, NW, Suite 900, Washington, DC 20036, Phone: (202) 827–7700 | Fax: (202) 355–9558, info@pcori.org.
Disclaimer All statements in this report, including its findings and conclusions, are solely those of the authors and do not necessarily represent the views of the Patient-Centred Outcomes Research Institute (PCORI), its Board of Governors or Methodology Committee.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.