Article Text

Abstract
Introduction Preclinical, clinical and epidemiological studies support the hypothesis that aberrant systemic metabolism of amyloid beta (Aβ) in the peripheral circulation is causally related to the development of Alzheimer’s disease (AD). Specifically, recent studies suggest that increased plasma concentrations of lipoprotein-Aβ compromise the brain microvasculature, resulting in extravasation and retention of the lipoprotein-Aβ moiety. The latter results in an inflammatory response and neurodegeneration ensues. Probucol, a historic cholesterol-lowering drug, has been shown in murine models to suppress lipoprotein-Aβ secretion, concomitant with maintaining blood–brain-barrier function, suppressing neurovascular inflammation and supporting cognitive function. This protocol details the probucol in Alzheimer’s study, a drug intervention trial investigating if probucol has potential to attenuate cognitive decline, delay brain atrophy and reduce cerebral amyloid burden in patients with mild-to-moderate AD.
Methods and analysis The study is a phase II, randomised, placebo-controlled, double-blind single-site clinical trial held in Perth, Australia. The target sample is 314 participants with mild-to-moderate AD. Participants will be recruited and randomised (1:1) to a 104-week intervention consisting of placebo induction for 2 weeks followed by 102 weeks of probucol (Lorelco) or placebo. The primary outcome is changed in cognitive performance determined via the Alzheimer’s Disease Assessment Scales-Cognitive Subscale test between baseline and 104 weeks. Secondary outcomes measures will be the change in brain structure and function, cerebral amyloid load, quality of life, and the safety and tolerability of Lorelco, after a 104week intervention.
Ethics and dissemination The study has been approved by the Bellberry Limited Human Research Ethics Committee (approval number: HREC2019-11-1063; Version 4, 6 October 2021). Informed consent will be obtained from participants prior to any study procedures being performed. The investigator group will disseminate study findings through peer-reviewed publications, key conferences and local stakeholder events.
Trial registration number Australian New Zealand Clinical Trials Registry (ACTRN12621000726853).
- neurology
- neurobiology
- dementia
- neuropathology
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first-in-human prospective randomised placebo-controlled study to assess the efficacy of probucol in delaying cognitive decline in individuals with mild cognitive impairment and mild-to-moderate dementia due to Alzheimer’s disease.
Strengths of the study methodology include randomisation of active/placebo allocation, the double-blinded nature of the study and the placebo-run in for the first 2 weeks of the study to monitor participant adherence to the treatment schedule.
The fixed-dose design may be a limitation of the study; further dose–response relationship studies may be indicated.
Introduction
Background and rationale
Alzheimer’s disease (AD) is a neurodegenerative disorder affecting approximately 50 million people worldwide. Extracellular deposition of amyloid beta (Aβ) is a hallmark pathological feature of AD featuring prominently within the hippocampal formation and entorhinal cortex. Amyloidosis is positively associated with cognitive decline in AD and targeting amyloidosis presently is a therapeutic priority.1 Recently, the US Food and Drug Administration Federal Drug Agency approved aducanumab, a treatment which decreases amyloid plaque burden in some patients with AD.2 3
Microvascular disturbances are the first pathological feature of AD that may include microbleeds; hypo-perfusion and or blood–brain barrier dysfunction with changes in the extracellular matrices associated with astrogliosis.4 5 Contemporary treatments for AD include cholinesterase inhibitors such as galantamine, rivastigmine or donepezil, to support synaptic activity, or memantine to regulate glutamate.6 Remarkably, of approximately 400 clinical trials in AD, to our knowledge, none to date have targeted the brain microvasculature, or peripheral metabolism of Aβ to reduce risk for, or progression of AD.
Systemic measures of soluble Aβ positively correlate with cerebral amyloidosis and loss of cognitive capacity in AD patients. Recent studies demonstrate striking evidence of the high predictive accuracy of plasma Aβ isoforms for individuals who progress to AD-dementia, decades before disease onset.7 8 While a causal association is suggested, the underlying mechanisms by which systemic Aβ metabolism may accelerate the risk of AD remain unclear. In humans, ~95% of soluble Aβ (Aβ1-40 and Aβ1-42 isoforms) is associated with plasma lipoproteins,9 primarily the triglyceride-rich lipoproteins of hepatically derived very-low-density-lipoproteins and of postprandial chylomicrons.10 To directly address the hypothesis of a lipoprotein-Aβ/capillary axis for AD, mice were engineered to synthesise human Aβ restricted exclusively to the liver to mimic peripheral lipoprotein-Aβ metabolism as seen in humans. Lam et al reported that aberrations in peripheral metabolism of lipoprotein-Aβ result in substantive neurodegenerative changes concomitant with loss of capillary integrity and function, blood-to-brain extravasation of lipoprotein-Aβ, marked neurovascular inflammation, and premature hippocampal-learning and memory deficits.11 Collectively, these findings provide a strong rationale to consider interventions that target and modulate peripheral metabolism of lipoprotein-Aβ to mitigate AD risk.
Probucol is a historic and safe cholesterol lowering drug, clinically used in Japan since 1985, with potent anti-inflammatory and antioxidant properties.12–14 Probucol was also shown to profoundly attenuate dietary induced synthesis and secretion of lipoprotein-Aβ15 16 concomitant with cerebral capillary integrity sparing.17 In a dietary-induced diabetic murine model, probucol was also found to support hippocampal-dependent memory recall.18 The pleiotropic properties of probucol and significant clinical use experience justifies considering repurposing probucol to test efficacy in supporting cognitive function in patients with AD.
We describe the probucol in Alzheimer’s study (PIA study). The study is a phase II placebo-controlled, double-blind clinical trial assessing the efficacy of probucol in AD. Key outcome measures include cognitive function, regional volumetric changes in brain and cerebral amyloid load.
Objectives
The primary objective of this study is to evaluate the efficacy of probucol (Lorelco) on cognitive performance in AD patients over a 104-week treatment period. The secondary objectives are (1) to evaluate regional volumetric changes and cerebral amyloid abundance in the brain of AD patients treated with probucol (Lorelco) over a 104-week treatment period, (2) to evaluate improvement or maintenance of quality-of-life parameters in patients with AD and (3) to assess the safety and tolerability of probucol (Lorelco) in patients with AD.
Methods and analysis
The methods reporting of this trial follow the recommendations of the Standard Protocol Items: Recommendations for Interventional Trials statement.19
Trial design
This is a single-site, phase II, randomised, double blind, placebo-controlled parallel group study in adults with mild-to-moderate AD. The study will assess the efficacy, safety and tolerability of the treatment of AD individuals with Lorelco. Participants, study doctors and researchers will be blinded to allocation of the study medication. The maximum study duration is 112 weeks (2 years, 8 weeks), with a treatment period of 104 weeks. There is a 4-week screening phase to ensure all participants have measurable mild or moderate AD and to ensure eligibility in the study. There is also a 4-week follow-up after the end of the treatment period. We aim to recruit 314 participants for this study. Eligible participants will be randomised in a 1:1 (active:placebo) ratio using permuted block randomisation. Eligible participants will be randomised to a unique participant code, which will be assigned to a participant number. Participant numbers will be randomised to either probucol (Lorelco) or placebo manufactured by Oxford Compounding. The trial coordinator will randomly assign the participant’s screening number to a unique participant code at week 1, day 1. Participants will be dosed as follows; week 1 and 2: 1× placebo taken in the morning, with food; week 3: 1×250 mg Lorelco (or matching placebo) taken in the morning, with food; and week 4–104: 1×250 mg Lorelco (or matching placebo) taken in the morning and in the evening, with food. An overview of the study design is shown in figure 1.
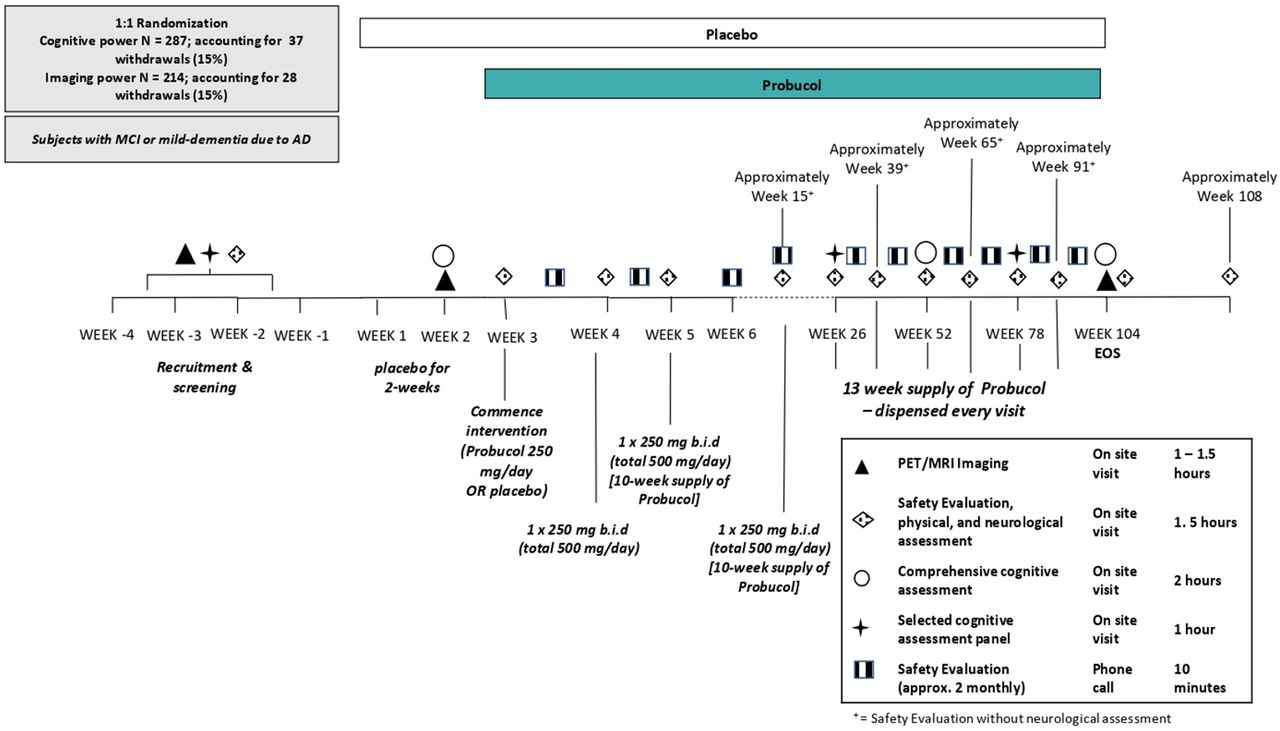
{kind=link}
Overview of probucol in Alzheimer’s study trial study design. AD, Alzheimer’s disease; EoS, end of study; MCI, mild cognitive impairment; PET, positron emission tomography; MRI, magnetic resonance imaging.
Study setting and recruitment
This study will be based in Australia. All assessments and blood collection will be completed at the Australian Alzheimer’s Research Foundation (AARF) based at Hollywood Specialist Centre, Nedlands, Western Australia. Positron emission tomography (PET) imaging will be completed at Sir Charles Gairdner Hospital, Nedlands, Western Australia. MRI will be completed at Envision Medical Imaging, Perth, Western Australia. To reach the targeted sample size, participants will be identified and recruited from the investigators’ private and public patient clinics; referrals from general practitioner or other specialists; phone calls to site via word of mouth; the Alzheimer’s Association Research Foundation website; or via database review where participants who have previously given permission for the site to contact them to inform them with new studies. Participants will also be recruited through advertising and editorials on social, print, radio and/or television media; correspondence and promotions via not-for-profit organisations and research partners; website and newsletter content.
Eligibility criteria
Individuals will be eligible for the study if they meet all of the inclusion criteria and do not satisfy any of the exclusion criteria listed in table 1.
Probucol in Alzheimer’s study trial inclusion and exclusion criteria
Intervention
Intervention description
During the initial recruitment and screening phase, patients will be screened against the exclusion criteria to ensure suitability for participation. During the screening phase, participants will undergo a short cognitive screening assessment, safety assessment, and complete a PET scan to determine cerebral amyloid load. Study medication will commence with a single dose escalation design with all participants receiving initially for 2 weeks 1× placebo consumed with food, after which baseline measures for cognitive performance, structural and functional brain MRI scans will be completed. Thereafter (week 3), patients receive for 1 week 1× Lorelco (250 mg) capsule (or matching placebo), taken with food. Commencing week 4, patients receive Lorelco 250 mg two times a day, consumed with food. Study medication will be dispensed at every study visit. Safety evaluations throughout the study will comprise physical/neurological examinations, ECG, vital sign measurements, standard laboratory tests and monitoring for AEs at weeks 3, 4, 5, 15, 26, 39, 52, 65, 78, 91, 104 and 108. At weeks 3, 4, 6, 20, 29, 47, 55, 73 and 81, the study coordinator will contact the patient or caregiver by phone record to determine any AEs.
Supply of study drug
Probucol used in this study will be commercially available tablets, Lorelco, produced by Aventis Pharmaceuticals and wholesaled by Otsuka Pharmaceutical Co. Lorelco tablets (250 mg) will be over-encapsulated inside an opaque capsule shell and backfilled with microcrystalline cellulose. Individual doses of Lorelco will be dispensed by the site pharmacy. Matching placebo opaque capsules with no active ingredients and a filler of microcrystalline cellulose will be compounded by Oxford Compounding.
Safety
Protocol violations should not lead to treatment discontinuation unless they pose a significant risk to participant safety. Trial stopping criteria, dose stopping rules and individual dosage adjustments are indicated in box 1.
Probucol in Alzheimer’s study trial stopping criteria and intervention dosage adjustments
Trial stopping criteria
Substantial deviations from the approved protocol.
Adverse-effects of unexpected type, severity or frequency are encountered.
As the trial progresses, the continuation of the trial would disadvantage some of the participants as determined by the investigator group, trial monitors or the data safety and monitoring committee.
*All participants will take part in a safety evaluation 4 weeks following discontinuation.
Dose stopping criteria and individual dosage adjustments
Dosing may be stopped or modified if suspected adverse drug reactions, changes in vital signs, ECGs or clinical laboratory results are observed, and these changes pose a significant health risk. Of particular note for dose discontinuation are the following criteria:
Corrected QT/QTc interval is in excess of 500 ms, or if it increases >50 ms compared with baseline (baseline will be calculated as the mean of two time points, screening and baseline day 1).
Occurrence of QT/QTc interval prolongation in association with symptoms of arrhythmia.
Subsequent prescription of a drug with potential QTc prolongation (antimalarial; macrolides; quinolones; triazole antifungals; antiarrhythmic; antiemetic; antidepressant; antipsychotics).
Evidence of liver dysfunction, or primary biliary cirrhosis.
If for any reason the investigator team determines that continued participation in the trial is not in the participant’s best interest.
Adverse events
The investigators will report any serious adverse events (AEs) occurring during the clinical trial, independent of direct causal relationship with the treatment, within 24 hours. Unblinding will be permissible in the event information is required to ensure the participants safety in case of an AE. The study doctor will provide Oxford Compounding with the unique participant code to unblind the participant. All AEs will be reviewed by the independent data safety and monitoring committee (DSMB), specifically appointed for the trial.
Participant withdrawal
If a participant decides to withdraw from the project, participants are asked to notify a member of the research team. If a participant withdraws consent during the research project, the study doctor and relevant project team members will not collect additional personal information from the participant. However, personal information already collected will be retained to ensure that the results of the research project can be measured properly and to comply with law. Participants will be made aware that data collected by the Sponsor up to the time the participant withdraws will form part of the research project results.
Adherence
The 2-week placebo period will serve to monitor participant adherence to the treatment schedule. Participants will also be required to return the study treatment webster packs at every clinic visit. In the event adherence falls below 80%, participants will be re-trained in the administration of the study medication. If adherence continues to be below 80% the participant will be withdrawn from the study.
Concomitant care
This study allows for ‘usual clinical care’. Some medications or treatments may not be permitted during participation in this study. The study doctor will collect information about concomitant medication use during every study visit and safety screening. Participants will not be permitted to take part in other studies/investigational treatments for AD or other health conditions while taking part in this study.
Study procedures
For the overall schedule of the trial and the exact timing of each procedure, refer to the Schedule of Assessments (table 2).
Overall Schedule of Assessments for the probucol in Alzheimer’s study trial
Screening assessments
Informed consent
The information and informed consent form (ICF) will be provided to patients at screening and signed consent must be provided prior to any study procedures being performed.
Medical history
A full medical history will be obtained at screening, including a detailed neurological history, other medical and surgical history, medication history and drug allergies. Demographic data including gender, ethnicity and race will be recorded.
Height and weight
Body height (cm) and weight (kg) will be measured, and body mass index will be calculated.
Pregnancy test
Female patients (women of childbearing potential only) will complete a urine human chorionic gonadotropin (hCG) pregnancy test at screening, baseline and again at end of study (EoS) visit.
Safety and tolerability assessments
Safety will be determined by evaluating physical and neurological examinations, vital signs, clinical laboratory parameters, 12-lead ECGs and AEs. Abnormal vital signs assessments, clinical laboratory safety tests, ECGs and physical examinations that are judged by the principle investigator as clinically significant will be recorded as AEs or serious adverse events (SAE). The timing of all safety assessments is presented in table 2.
Vital signs
Vital signs assessments will include systolic and diastolic blood pressure, heart rate (HR), respiratory rate (RR) and body temperature. Patients should be resting in a supine position for at least 5 min prior to and during vital signs measurements.
Clinical laboratory safety tests
Fasted blood samples (minimum 8 hours fast) will be collected by venepuncture at screening, baseline, weeks 3, 4, 5, 15, 26, 39, 52, 65, 78, 91 and 104 weeks (an estimated 13 mL of blood will be collected per visit and a total of 160 mL will be collected over the 2-year study).
Blood samples for haematology, serum biochemistry (including liver function tests) will be collected at selected time points throughout the study (see table 2). Test results will be monitored for potential AEs, including gastrointestinal bleeding (haemoglobin) and rhabdomyolysis (plasma CK). Apolipoprotein E genotype will also be determined.
ECG
Twelve-lead ECGs will be assessed (including but not limited to the measurements of ventricular HR, PR interval, RR interval, QRS duration, QT interval and QTcF). Screening and prior to first dose, triplicate 12-lead ECGs (collected within 5 min with each reading separated by at least 1 min) will be taken to establish eligibility at baseline. Triplicate ECGs will also be recorded at the EoS visit. The average value for the triplicate will be used for assessing QTcF inclusion criteria. All other ECGs will be single readings.
ECG normal ranges are as follows: PR interval: 120 ms–220 ms (inclusive); QRS duration: <120 ms; QTcF ≤450 ms (males); QTcF ≤460 ms (females); HR 45–100 beats/min (inclusive).
Physical examination
A full physical examination will be performed at screening and at the EoS visit. The full physical examination will include, at a minimum, assessment of the following systems: skin, head, ears, eyes, nose and throat, lymph nodes, heart, chest, abdomen and extremities, and a neurological examination (assessment of speech, cranial nerves, peripheral nerves, motor power, deep tendon reflexes, sensation, coordination and gait) and any other focused assessments suggested by the presence of specific symptoms. All other scheduled assessments will be symptom-directed.
Cognitive screening assessments
Free and Cued Selective Reminding Test
The Free and Cued Selective Reminding Test (FCSRT)20 assessment will be performed at screening only for eligibility determination. A cueing index of ≤0.79 is required for study entry. The FCSRT is a cued recall test that uses a controlled encoding technique to ascertain that impairment in recall and cueing are due to a memory deficit rather than a failure of encoding.
Clinical Dementia Rating scale
The Clinical Dementia Rating (CDR)21 provides two scores, a global score (GS) and a sum of boxes (SOB). The GS distinguishes a participant’s level of impairment into the following categories: 0 (normal); 0.5 (questionable dementia; 1 (mild dementia); 2 (moderate dementia) and 3 (severe dementia). The SOB is scored from 0 to 18 with higher scores indicating a greater level of impairment. The scale covers six domains: memory, orientation, judgement and problem solving, community affairs, home and hobbies and personal care.
Mini-Mental State Examination
The Mini-Mental State Examination (MMSE)22 is a brief, widely used 30-item assessment of global cognition examining orientation, registration, calculation, recall, attention and language. The spelling of WORLD backwards will not be used in this protocol. Participants who score <22 at screening will be ineligible for study entry. MMSE will be assessed at screening and EoS.
All cognitive assessments will be performed by an independent assessor who will be blinded to treatment allocation.
Outcome measures
Primary outcome measures
The primary outcome measure will be the change in the Alzheimer’s Disease Assessment Scales-Cognitive Subscale test (ADAS-Cog).23 The ADAS-Cog is the most widely used test to measure cognition in RCT’s for AD. The ADAS-Cog consists of the following tasks: Word Recall Task; Following Commands; Constructional Praxis; Delayed Word Recall; Naming Objects and Fingers; Ideational Praxis; Orientation; Word Recognition; Spoken Language; Comprehension and Word Finding Difficulty. The ADAS-Cog 12 has a total scoring range of 0–70, where 70 represents the most severe impairment and 0 represents the least impairment. The full suite of ADAS-Cog will be assessed at baseline, 26, 52, 78 and 104 weeks. At the discretion of the study sponsor, the extended ADAS-Cog, with the additional tasks Maze and Number Cancellation, may also be administered.
Secondary outcomes
A secondary outcome measure will be assessment of brain morphometry and volume determined via MRI at baseline (preintervention) and EoS. The MRI protocol will include the acquisition of three sets of data (1) Volumetric isotropic T1 scan (6.5 min). This will allow voxel wise segmentation and volumetric analyses (eg, grey matter volume) to assess volume changes in characteristic locations which can yield diagnostic accuracy on approximately 90%.24 Mesial temporal lobe (hippocampus and entorhinal cortex via Scheltens grading), global cortical atrophy (Pasquier scale) and parietal atrophy (Koedam score) as well as inferior lateral ventricle size will be assessed. Brain volume indices indicate that patients with AD have accelerated rates of brain volume loss of up to 4.5% per year compared with normal controls (1%). (2) 3D FLAIR scan (3.5 min). This will demonstrate the small vessel ischaemic lesion load which will be scored according to the Fazekas method.25 (3) Susceptibility weighted imaging (SWI)—a means of measuring micro bleed load and indicator of amyloid angiopathy and for the purposes of Quantitative Susceptibility Mapping (~7 min).25 This method quantitates regional brain iron content which is altered in AD compared with normal controls. Mesial temporal, basal ganglia, cingulate, cortical region of interest comparisons will be performed at baseline and at treatment completion. Visible micro bleeds on the SWI will be graded using the Brain Observer Micro-Bleed Scale.26 27
Cerebral amyloid load will be assessed as an additional imaging outcome measure. Brain amyloid imaging will be done with amyloid tracer PET scans at baseline and at EoS. Dynamic and static PET imaging will be acquired. Analysis will include (1) visual assessment of amyloid load, (2) quantitative assessment of amyloid burden, including standard uptake value ratio and (3) dynamic imaging for blood perfusion measures.
Quality of life will be assessed as a secondary outcome measure via the Alzheimer’s Disease Co-operative Study Mild Cognitive Impairment Activities of Daily Living (ADCS-MCI-ADL24).28 The ADCS-MCI-ADL24 is a study partner 24-item questionnaire evaluating perceived difficulties with functioning in several activities of daily living across a variety of domains. The Depression Anxiety Stress Scale (DASS-21), a self-report 21-item will be used to determine levels of depression, anxiety and stress.29 Participants are read a statement and asked to rate each statement on a 4-point scale as to how much it relates to them. DASS-21 will be administered at day 1 /week 1, week 26 and EoS.
Statistics
Estimated sample size and power
Estimated sample sizes are calculated for the two outcome measurements: ADAS-Cog and grey matter atrophy (hippocampal). In order to ensure that the study has sufficient power to detect differences in both of the primary outcomes, the sample size chosen is the maximum of that calculated for each primary outcome.
The primary analysis is an intention-to-treat analysis and will include all randomised participants. Data will be analysed using both generalised estimating equations (GEE) and Bayesian analysis. The analysis of primary endpoints will use linear mixed-effects models, with random slopes and intercepts. For the ADAS-Cog, using mixed model analysis published estimates from the Alzheimer’s disease neuroimaging initiative (ADNI) cohort30 suggest a sample size of 125 AD participants per trial arm (total N=250) will be required for power at 0.8 to detect a drug effect of 25% over 2 years and assuming a decline from baseline of 1.10 standardised units on the composite (SD change=0.83). For the MRI markers, Ledig et al reported the sample sizes required for a 25% intervention reduction over 2 years based on 322 patients with AD (with 117 followed for 24 months) and a reduction of 10.2% (6.2) for hippocampus.31 Sample size calculations based on hippocampal volume suggest that 93 participants per treatment arm are required (total N=186). Assuming a 20% attrition rate, a sample of 314 individuals will be recruited for the cognitive study, and 233 individuals will be randomly chosen for the imaging study.
Statistical analysis
Outcomes
The primary analysis is an intention-to-treat analysis of all randomised participants. Data will be analysed using both GEE and Bayesian analysis. The analysis of primary endpoints will use linear mixed-effects models, with random slopes and intercepts. Analysis of all primary and secondary endpoints contrasting probucol and placebo, after adjusting for covariates, will use mixed effects-regression with ‘random’ intercepts and slopes (as has been used for power calculations). Mean differences and associated 95% CIs will be presented for the ‘fixed’ effect of probucol treatment. No formal interim analyses are planned at this time.
Additional analyses
Further analysis investigating the relationship between change scores (postintervention minus preintervention scores) for the primary ADAS-Cog with MRI volumes (total grey matter, hippocampus and medial temporal lobe volumes) and specific blood biomarkers (eg, plasma lipoprotein-Aβ) will be considered using Pearson (or Spearman where appropriate) correlation analysis. For all other correlations between recorded variables that lack an a priori hypothesis, control of statistical errors will be carried out using Holm-Sidak corrections for multiple comparisons.
If probucol treatment is successful, a directed acyclic graph Bayesian network analysis will be carried out a posteriori on variables identified to be significant predictors of either grey matter arrest or neuropsychological performance to better elucidate mechanisms of the effect of probucol. Greedy equivalence search will be used to identify statistical conditional dependencies between variables and directionality will be estimated using the linear, non-Gaussian, acyclic causal models approach.32 33 Goodness of fit will be estimated using a χ2 test contrasting the identified model against a saturated model.
In addition to Bayesian analyses, the traditional general linear model analysis will also be used to compare probucol to placebo, after adjusting for covariates. The GEE method, which extends the generalised linear model to allow for analysis of repeated measurements or other correlated observations, will also be used. Missing data on the ADAS-Cog will not require data imputation. The scoring methodology for the ADAS-Cog as proposed by Verma et al will be used as it estimates cognitive impairment using the set of items answered by the patients.23 Any additional missing data will be identified using missing values analysis and will be replaced using multiple imputation where appropriate. Mean difference and associated 95% CIs will be presented. Data will be analysed using Stata V.16.
Data and safety monitoring
Data management
Data will be collected by study delegated personnel on paper source maintained in a participant study binder in secure facilities at AARF. Identifiable data will be stored securely and kept in a locked cabinet with access restricted to the investigator team, site and monitoring personnel. All other data will be de-identified to sure confidentiality of participant data. Data will be stored electronically on password protected web-enabled clinical trial data electronic management system (REDCap) located in an ISO27001 compliant facility at Curtin University. Clinical records collected at recruitment will be kept in a locked cabinet in a locked office at the site and will collectively be housed in secure facilities at the AARF. Participants’ study information will not be released outside of the study without the written permission of the participant. All data will be securely archived as per the Sponsor’s data policy for a minimum of 25 years.
Trial monitoring and formal committees
The trial monitoring committee, comprised the principal investigator, key trial staff including the trial manager, a nurse representative and a consumer representative, are responsible for trial setup, ongoing management and promotion of the trial. The trial steering committee (TSC), comprising the investigator team including geriatricians, cardiologists, neuroradiologists, nuclear medicine physicians, neuropsychologists, a biostatistician, clinical biochemists, consumer and community representatives, will provide overall supervision of the study and are responsible for interpretation and dissemination of results.
AARF employs independent data auditors who will monitor and audit compliance of data entry/management, legislation, regulations, guidelines and codes of practice, at quarterly intervals. Findings from each audit will be discussed with the study coordinator and thereafter with the investigator team to ensure any action items are addressed promptly and appropriately.
An independent DSMB will oversee the safety aspects of the study. The DSMB consists of members with expertise in clinical pharmacology, biostatistics, clinical trial design and clinical cardiology. Members of the DSMB will not be investigators of the study nor will they have any conflict of interest with the investigators. The committee will meet periodically to advise the TSC on the progress of efficacy and safety data as it accumulates throughout the course of the study. The TSC and DSMB will provide independent oversight of the study.
Patient and public involvement
The study was developed in consultation with a consumer advocate representing the Consumer and Community Involvement Programme (https://cciprogram.org/). The trial will be overseen by the TSC, including patient and public members.
Ethics and dissemination
Ethics approval
The PIA study has been approved by Bellberry Ltd Human Research Ethics Committee (HREC2019-11-1063; refer to online supplemental file 1 for the approved study protocol). This trial is registered in accord with the WHO Trial Registration Data Set. The Universal Trial Number is U1111-1259-0486. Where applicable, approved protocol amendments will be communicated to the trial personnel and relevant committees.
Supplemental material
Informed consent and withdrawal from the study
Participants will be in the mild stages of dementia and therefore are expected to be able to provide informed consent. An investigator or research staff delegate will explain all study procedures and possible risks to the participant. The participant and their nominated study partner will have an opportunity to have all questions answered and thereafter will sign and date the ICF, indicating willingness to participate in the study with an option to consent for blood samples stored and used for future research purposes (online supplemental appendices A–D). Participants will be informed prior to consent that they can withdraw at any time without their care being affected in any way.
Supplemental material
Dissemination
Participants will be updated about the progress and results of the study via presentations or newsletters from the investigator group. The results will be disseminated via peer-reviewed publications, key conferences and local stakeholder events, under the Consolidated Standards of Reporting Trials guidelines.34
Trial status
This study is in the process of recruiting participants and expected to complete in 2026.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @profcmreid, @Robinsonsuz, @Ryu_Takechi
Contributors VL, RC, RF, MB, SSD, RT and JM conceived the study concept. VL, RC, RF, MB, GW, LF, SSD, EC, RT and JM designed the trial protocol. LF, CFO, PL, NL, CMR, JKF, SR, MV and BH commented on the methods and contributed to the development of the study. VL, EC and JM drafted the manuscript. All authors revised the manuscript and approved the final version.
Funding The PIA study is funded by the National Health and Medical Research Council Medical Research Future Fund for Neurological Disorders (MRF1201204), the Multiple Sclerosis Society of Western Australia (MSWA) and the McCusker Charitable Foundation.
Disclaimer Curtin University, the National Health and Medical Research Council and MSWA do not have any responsibility relating to study design; collection, management, analysis and interpretation of data; writing of the report; and the decision to submit the report for publication.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.