Article Text

Abstract
Introduction The efficacy and safety of anticoagulant treatment is not established for patients with acute symptomatic isolated distal deep vein thrombosis (IDDVT). In real-world clinical practice, both therapeutic and prophylactic anticoagulation are used for acute IDDVT. However, therapeutic anticoagulation is associated with higher risk of bleeding than prophylactic anticoagulation. Thus, this study aims to assess the efficacy and safety in patients with first acute symptomatic IDDVT treated with therapeutic or prophylactic anticoagulation using rivaroxaban.
Methods and analysis This study is a prospective, multicentre, single-blind, randomised controlled trial. Outpatients with a first, acute, symptomatic, objectively confirmed IDDVT in four centres from 1 August 2021 are recruited. Eligible patients are randomised in a 1:1 ratio to receive prophylactic anticoagulation (rivaroxaban 10 mg once a day for 3 months) or therapeutic anticoagulation (rivaroxaban 20 mg once a day for 3 months). All patients are followed for 6 months. The primary efficacy outcome is radiographically confirmed recurrent venous thromboembolism. The primary safety outcome is the incidence of major or clinically relevant non-major bleeding events.
Ethics and dissemination This study has been approved by the Ethics Committee of Zhongshan Hospital Fudan University (B2021-175R). Study results will be disseminated through peer-reviewed journals.
Trial registration number NCT04967573
- vascular surgery
- anticoagulation
- thromboembolism
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study is the first prospective, multicentre, randomised controlled trial comparing the efficacy and safety of therapeutic and prophylactic anticoagulant therapy in patients with first acute symptomatic isolated distal deep vein thrombosis (IDDVT).
Due to the high negative predictive value, D-dimer is taken as an adjunctive marker to rule out DVT in this study.
Only one kind of direct oral anticoagulant, rivaroxaban, is used to ensure the comparability between therapeutic and prophylactic anticoagulant therapy.
This study focuses on Asians resulting in regional limitations.
Introduction
Isolated distal deep vein thrombosis (IDDVT) is a common disorder that involves the infrapopliteal veins, including the peroneal, anterior tibial, posterior tibial veins and the muscular veins.1 According to the report from the International Society on Thrombosis and Haemostasis, the incidence of venous thromboembolism (VTE) is 115–269 per 100 000 population worldwide, IDDVT represents 20%–50% of all lower-limb DVTs.2 3 There is growing evidence that the risk factors of IDDVT are different from those of pulmonary embolism (PE) and proximal DVT.4 An international, perspective registry revealed that IDDVT was more associated with transient risk factors such as recent surgery or travel, hospitalisation, whereas proximal DVT was more prevalent in elderly patients, pregnant women and chronic states, like active cancers.5 However, if untreated, IDDVT might propagate to proximal veins and even cause PE. Thus, it is a clinically important issue about how to treat IDDVT.
Currently, the optimal management of IDDVT in terms of the need for anticoagulant treatment, the type and duration of anticoagulant treatment remains undefined. A recent meta-analysis enrolled 8 randomised controlled trials (RCTs) reporting on 1239 participants. Of them, five RCTs determined that compared with no intervention or placebo for IDDVT, anticoagulation for up to 3 months reduced the risk of recurrent VTE (Risk Ratio (RR) 0.34; 95% CI 0.15 to 0.77) and did not increase the risk of major bleeding (RR 0.76; 95% CI 0.13 to 4.62). Furthermore, the other three RCTs confirmed that anticoagulation with a vitamin K antagonist over 3 months was associated with lower risk of recurrent VTE (RR 0.42; 95% CI 0.26 to 0.68) compared with anticoagulation for 6 weeks. The extended duration of anticoagulant treatment did not increase the incidence of major or clinically relevant non-major bleeding events.6 Another meta-analysis also suggested that anticoagulant treatment over 6 weeks was better than a shorter duration, as a longer course was associated with a lower rate of recurrent VTE and proximal extension.7 The GARFIELD-VTE registry reveals a global trend that most patients with IDDVT are given anticoagulant therapy.4 Moreover, compared with warfarin, direct oral anticoagulant (DOAC) was observed with lower VTE recurrence, major bleeding, and mortality in both the IDDVT and proximal DVT groups.8 The RIETE registry and the RE-COVERY DVT/PE study reported that more than half of patients with IDDVT received DOACs in clinical practice.9 10 Overall, the 2016 American College of Chest Physician guidelines suggest the same anticoagulant treatment (at least 3 months) for acute high-risk IDDVT as for acute proximal DVT.11
As to the dose of anticoagulant therapy for IDDVT, Yoon et al reported significantly higher incidence of VTE complications in IDDVT patients treated with prophylactic anticoagulation (30%) than in those treated with therapeutic anticoagulation (10%) (p=0.0003).12 Another single-centre retrospective study also confirmed that therapeutic anticoagulation was associated with a lower rate of proximal DVT or PE after isolated calf DVT (OR 0.33; 95% CI 0.12 to 0.87), but an increased risk of bleeding events (OR 4.87; 95% CI 1.37 to 17.3).13 However, Dentali et al retrospectively analysed 308 patients with IDDVT and active cancer and found that there is no significant difference in the rate of recurrent VTE during and after treatment among therapeutic, intermediate and prophylactic anticoagulation groups.14 Considering the conflicting results and the frequency of IDDVT, a prospective, multicentre RCT is warranted to investigate the optimal intensity of anticoagulant treatment for patients with IDDVT.
Objectives
The purpose of this study is to compare the efficacy and safety of prophylactic or therapeutic anticoagulant therapy with rivaroxaban in patients with first acute symptomatic IDDVT.
Methods and analysis
Study design
This is a prospective, multicentre, single-blind, RCT, which complies with the Standard Protocol Items: Recommendations for Interventional Trials guidelines.15 Outpatients with a first, acute, symptomatic, objectively confirmed IDDVT will be invited to participate in this study. The diagnosis of IDDVT is established by symptoms, signs, aberrant D-dimer value and an incompressible venous segment in infrapopliteal veins on compression ultrasonography. The enrolled patients will be randomly assigned in a 1:1 ratio to prophylactic or therapeutic anticoagulation. Random allocation of each patient was centrally determined by the Department of Vascular Surgery, Zhongshan Hospital Fudan University. Using a web-based interface, the investigator received by smartphone the number and the corresponding group of the patient. All patients and outcome evaluators are blinded to group assignment.
This work was initiated by Zhongshan Hospital Fudan University in early 2021. Three additional sites have been invited to join this study, including Shanghai General Hospital, Huadong Hospital affiliated to Fudan University and Shanghai Wusong Hospital . All study site investigators, research staff have been provided with the protocol and guidance documents to ensure the homogeneity. Additionally, the principal investigator met and delivered a lecture to introduce the protocol to the site investigators.
Participants
When potential participants meet the inclusion criteria, they are referred to the study by the site investigator. Patients who give the written informed consent are further assessed by compression ultrasonography, blood pressure measurement, blood routine examination, liver function, renal function and coagulation function tests for eligibility based on exclusion criteria. Then, they will be enrolled in this study.
Criteria
Inclusion criteria
Patients aged 18–90.
Outpatients with a first, acute (within 2 weeks), symptomatic, objectively confirmed IDDVT.
Compliance to the scheduled follow-up plan.
Ability and willing to participate and sign the informed consent.
Exclusion criteria
Any absolute contraindication to anticoagulant treatment.
Pregnancy or breast feeding.
Systolic pressure >180 mm Hg or diastolic pressure >100 mm Hg.
Platelet count <100×10⁹/L.
Serum creatinine >180 mmol/L or creatinine clearance ≤30 mL/min.
Liver disease associated with coagulopathy and high risk of bleeding.
Clinically suspected or confirmed PE.
Ipsilateral or contralateral proximal DVT.
Any indication for long-term anticoagulation.
Enrolled in another clinical trial simultaneously.
Life expectancy <6 months.
Withdraw criteria
Patients voluntarily request to withdraw.
Investigators consider it necessary for discontinuation of the study medication from a professional view.
Study procedures
The general clinical characteristics, onset time, symptoms, signs, detailed description of the ultrasound results, D-dimer value and related risk factors of enrolled patients are recorded using a standard case report form (CRF) at the time of recruitment. Patients who are assigned to the prophylactic anticoagulation group will receive oral rivaroxaban 10 mg and a matching rivaroxaban placebo once a day for 3 months. In contrast, patients in the therapeutic anticoagulation group will receive two rivaroxaban 10 mg orally once a day for 3 months. The study drug is discontinued after 3 months. The clinical surveillances are conducted by phone at 2 weeks, by in-person at 6 weeks and 3 months. At each contact, the trained research staff will record the drug adherence, symptoms, suspected recurrent VTE and bleeding events. Additionally, two in-person follow-up visits also comprise a D-dimer test and a bilateral whole-leg compression ultrasonography. Finally, a follow-up interview by phone is done 6 months after inclusion in this study. The study procedure is showed in figure 1.

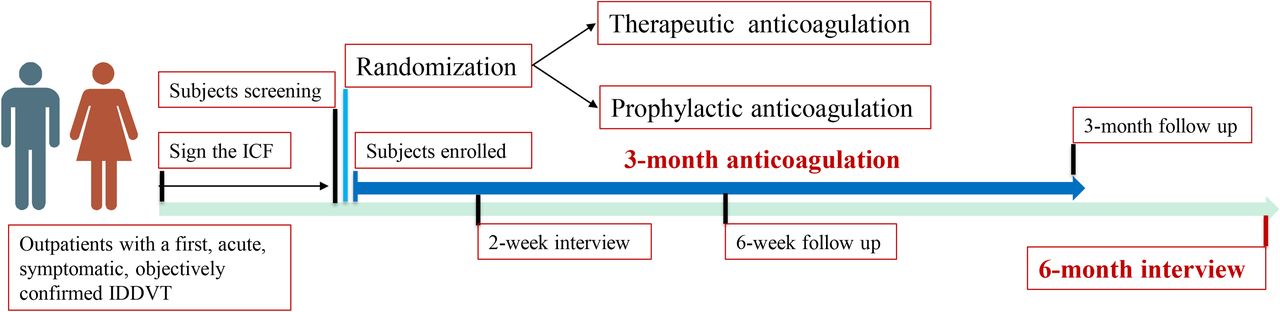{kind=link}
Schematic flow chart of the study. ICF, informed consent form; IDDVT, isolated distal deep vein thrombosis.
Patients are instructed to contact the research staff at any time in case of any bleeding events, new or worsening VTE symptoms. If the participants require open surgery, rivaroxaban should be stopped at least 24 hours before the elective procedure, or immediately for emergent settings. If the discontinuation of rivaroxaban >7 days for any reason or uncontemplated pregnancy, the patients should withdraw from the study. If the participants report potential major bleeding events, they are asked to have a detailed assessment in the department of emergency.
Outcome measures
The primary efficacy outcome is radiographically confirmed recurrent VTE, including proximal extension of IDDVT, new contralateral proximal DVT and PE at 6 months after randomisation. Of them, proximal extension and new contralateral proximal DVT are defined as ultrasound-proven incompressible venous segment in the popliteal, femoral veins and positive findings of iliac and cava veins on spiral CT (venogram) or venography. The diagnosis of PE relies on clinically suspicion and further chest computed tomographic angiography, ventilation perfusion lung scan or pulmonary angiogram.
The primary safety outcome is the major or clinically relevant non-major bleeding events at 3 months after the initiation of treatment. Major bleeding events are defined according to the International Society on Thrombosis and Haemostasis criteria.16 Clinically relevant non-major bleeding events refer to which do not meet the mentioned criteria of major bleeding but need at least an unscheduled in-person consultant. Secondary outcomes include individual component of primary outcomes (such as proximal extension, major bleeding, etc), residual vein occlusion, postthrombotic syndrome and all-cause death at 6 months after randomisation. All uncertain outcomes should be judged by a central adjudication committee whose members are blinded to the study design.
Data collection
Researchers use a standardised CRF to collect information from the participants. Data include the general clinical characteristics (age, gender, body mass index, race), onset time, symptoms, signs, risk factors for VTE (recent surgery of trauma, pregnancy, oestrogen use, active cancer, varicose veins, history of VTE, ambulatory status), detailed description of the ultrasound results (involved venous segment, length of the thrombus), laboratory tests (especially for the coagulation function), drug adherence, subsequent testing for VTE, recurrent VTE, bleeding events and all-cause death within 6 months. The collected data will be uploaded into an electronic data capture system.
Sample size and statistical analysis
Based on a previous retrospective study and the current experience, the estimated rate of recurrent VTE was 10% and 3% in prophylactic and therapeutic anticoagulation group, respectively.13 According to statistical requirements, we used 80% targeted power at the α level of 0.05 for a two-sided test. Sample size of 191 in each group was thus obtained. Besides, for the primary safety outcome, a sample size of 163 in each group was developed with 80% power at a two-sided 5% level of significance, assuming that the incidence of bleeding events is 2% and 9% in prophylactic and therapeutic anticoagulation group, respectively.13 Moreover, considering the dropout rate of 20%, a total of 480 patients are required for this study eventually.
Primary outcome analyses are performed in both intention-to-treat (ITT) and per-protocol (PP) modalities to ensure the results robust. In the ITT analysis, all enrolled patients will be analysed as for the group they are initially allocated to. However, patients with regimen violations will be excluded from the PP analysis. The rate of recurrent VTE at 6 months following randomisation and its 95% CI will be generated and compared between the two groups using a Fisher exact test or χ² as appropriate. For the primary safety outcome, the same statistical analysis will be applied. Moreover, sensitivity analysis will be conducted to evaluate the robustness of the primary results. All the data will be analysed with STATA software (V.15).
Data management
During the study, anonymised data will be stored in a secure manner according to the data management plan. No interim analysis is planned. After all the data uploaded into the electronic data capture system, the principal investigator, data manager and statistical scientists will audit data. Then, all data will be locked and can not be edited. Access will only be authorised to the statistical scientists and only be used for the planned statistical analysis.
Patient and public involvement
Patients or the public are not involved in the design, recruitment, or conduct of this research. Research results will be disseminated to the patients or the public through medical journals.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors MZ, WZ and YZ are joint first authors. ZS obtained funding. MZ, JM, TX and ZS designed the study. MZ, WZ and YZ drafted the manuscript. MZ and TX registered the study and developed the CRF. JM and ZS contributed to the critical revision of the manuscript for important intellectual content and approved the final version of the manuscript. All authors read and approved the final manuscript. JM and ZS are the study guarantors.
Funding Clinical Research Project of Zhongshan Hospital (No. 2020ZSLC28), Fudan University.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.