Article Text

Abstract
Introduction Anaemia is highly prevalent in critical illness and is associated with impaired outcomes during and after hospitalisation. However, the impact of interventions designed to attenuate or treat anaemia during critical illness on post-hospitalisation haemoglobin recovery and functional outcomes is unclear.
Methods and analysis The Practical Anemia Bundle for Sustained Blood Recovery (PABST-BR) clinical trial is a pragmatic, open-label, parallel group, single-centre, randomised clinical trial assessing the impact of a multifaceted anaemia prevention and treatment strategy versus standard care for improvement of haemoglobin concentrations and functional outcomes after critical illness. The intervention, which will be delivered early in critical illness for those with moderate-to-severe anaemia (ie, haemoglobin <100 g/L), includes three components: (1) optimised phlebotomy, (2) clinical decision support and (3) pharmacological anaemia treatment directed at the underlying aetiology of anaemia. In-person assessments will occur at 1 and 3 months post-hospitalisation for laboratory evaluations and multidimensional functional outcome assessments. The primary outcome is differences in haemoglobin concentrations between groups, with secondary endpoints of anaemia-related fatigue, physical function, cognition, mental health, quality of life, phlebotomy volumes and frequency, transfusions, readmissions and mortality through 1-year post-hospitalisation.
Ethics and dissemination The study has been approved by the Institutional Review Board of the Mayo Clinic in Minnesota, USA. A Data Safety Monitoring Plan has been created in accordance with the policies of the Institutional Review Board and the study funder, the National Heart, Lung and Blood Institute of the National Institutes of Health (NIH). The study will comply with NIH data sharing and dissemination policies. Results will be presented at national and international meetings and published in peer-reviewed journals. Designing and testing strategies to optimise haemoglobin recovery and improve functional outcomes after critical illness remain important research gaps. The PABST-BR trial will inform the development of a larger multicentre clinical trial.
Trial registration number NCT05167734.
- anaemia
- adult intensive & critical care
- blood bank & transfusion medicine
- intensive & critical care
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study tests a novel multifaceted bundle of interventions to attenuate the severity of anaemia and promote haemoglobin recovery early in the course of critical illness. By including strategies to both attenuate and treat anaemia, a more substantial impact on haemoglobin recovery may be observed than what would be experienced with any intervention in isolation.
Pharmacological treatments of anaemia are targeted to the underlying aetiology of anaemia, which may enhance both safety and efficacy of the intervention.
The trial includes multiple important secondary outcomes, including those related to multidimensional post-hospitalisation functional outcomes that may be negatively impacted by anaemia, including physical function, cognition, mental health, quality of life and fatigue.
Limitations include an open label design and anticipated enrolment numbers which are not powered for secondary outcomes. However, this study will assess feasibility of the study intervention and provide safety and efficacy signals to inform a future multicentre trial.
Introduction
Anaemia is common in the critically ill and associated with fewer days alive and at home,1 impaired physical function2 and decreased survival in the first year after hospitalisation.3 As up to 50% of critical illness survivors have substantial functional deficits in one or multiple domains related to physical function, cognition, mental health and quality of life,4 it is paramount to identify modifiable risk factors for impaired functional recovery, of which anaemia may be one potential target. Recent data from survivors of critical illness suggests that anaemia during critical illness is associated with impaired physical function 3 months later.2 Additional evidence suggests that survivors with improved early post-hospitalisation haemoglobin recovery are less likely to be rehospitalised or die in the subsequent year.5
Numerous clinical trials in critically ill patients have evaluated treatment of anaemia with iron and/or erythropoietin (EPO).6–11 While these therapies have consistently augmented haemoglobin recovery, they have had inconsistent results on transfusion reductions and mortality and have not been widely adopted into clinical practice. However, there is increasing recognition that anaemia may contribute to persistent functional impairments after survival of hospitalisation.1 2 5 As such, strategies to augment haemoglobin recovery may favourably influence post-hospitalisation outcomes. Two recent trials suggest that intravenous iron, with or without EPO, may improve haemoglobin recovery after critical illness, with one trial showing lower 90-day mortality and the other showing reduced readmissions (secondary outcomes).10 12 Importantly, anaemia treatments are typically delivered late in the disease process after anaemia has fully developed and are generally not targeted to the underlying aetiology of anaemia. Fortunately, there are strategies available that may prevent or attenuate the severity of anaemia development such as minimising iatrogenic blood loss via phlebotomy,13 ensuring appropriate bleeding prophylaxis for high-risk patients and preventing haemodilution. Further, the use of real-time clinical decision support may assist in these efforts, though this has not been formally studied in critically ill patients.
In this investigation, we describe the protocol for a pilot randomised clinical trial of a novel multifaceted anaemia prevention and treatment bundle for critically ill adults with anaemia. These results will be used to assess feasibility, refine the intervention and inform a larger multicentre clinical trial.
Methods and analysis
Study design
This is a pragmatic, open-label, parallel group, single-centre, randomised clinical trial for superiority assessing the impact of a multifaceted anaemia prevention and treatment strategy versus standard care for improvement of haemoglobin concentrations and functional outcomes after hospitalisation for critical illness. The trial titled the ‘Practical Anemia Bundle for Sustained Blood Recovery (PABST-BR)’ has been registered on ClinicalTrials.gov. It has been approved by the appropriate Institutional Review Board (IRB) at the Mayo Clinic (Minnesota, USA). Trial enrolment is anticipated between March 2022 and March 2024.
Study population
Critically ill patients will be enrolled using the following inclusion criteria: age >18 years, intensive care unit (ICU) admission in an adult ICU at the study institution (online supplemental table 1), haemoglobin <100 g/L, anticipated ICU length of stay >48 hours after enrolment, current ICU duration <7 days and local residence to facilitate follow-up assessments after hospitalisation. Patients meeting these inclusion criteria without exclusions (table 1), or their legal proxies, will be approached by trained study coordinators for written informed consent using a study-specific consent form (online supplemental content).
Supplemental material
Exclusion criteria for study enrolment
Randomisation
Eligible participants will be approached by trained study personnel and randomised 1:1 to active intervention versus standard care (ie, control) using a stratified permuted block design by anaemia aetiology (ie, iron-responsive vs inflammatory) and ICU admission indication (ie, surgical vs non-surgical). Randomisation will be performed through the Research Electronic Data Capture (REDCap) randomisation module.
Study intervention
The intervention arm is multifaceted with three primary components (figure 1): (1) Optimised phlebotomy, defined by default microvolume blood collections (ie, 0.1–1 mL vs standard 3–10 mL per laboratory order), closed loop blood sampling (ie, return of blood waste for phlebotomy draws from existing blood catheters) and bundled laboratory timings, all performed by a dedicated phlebotomy team independent from the treatment team; (2) Clinical Decision Support, including bedside and electronic visual aids (online supplemental figure 1) and best practice alerts embedded in the electronic health record (EHR; online supplemental figure 2) and a daily rounding checklist (figure 2) reminding the care team to minimise non-essential laboratory testing, mitigate patient-specific bleeding risk, eliminate unnecessary intravenous fluids and ensure adequate nutritional status; and (3) Pharmacological anaemia treatment, administered immediately following enrolment, targeted to the aetiology of anaemia, including (1) anaemias responsive to iron supplementation alone (ie, anaemia secondary to acute blood loss or with laboratory evidence of iron deficiency or low iron stores (ie, ferritin <100 ng/mL or transferrin saturation <20%)), and (2) anaemias of inflammation requiring erythropoietic stimulation (ie, anaemias occurring secondary to underlying medical illness in the absence of iron deficiency or pre-existing anaemia of other causes (eg, thalassaemia, sickle cell disease)). Anaemia aetiology will be determined on review of laboratory values and clinical history with treatment decisions adjudicated pre-randomisation.
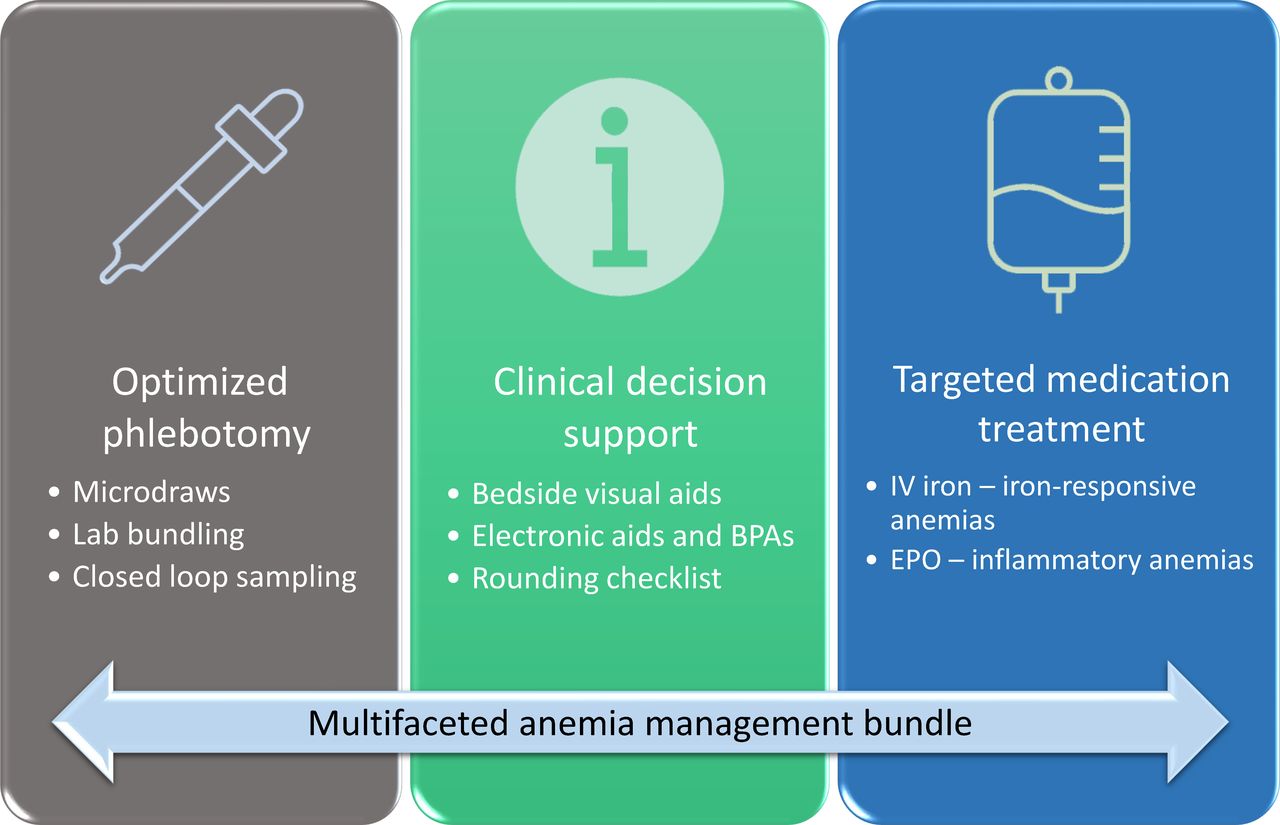
Anaemia management bundle for patients in the intervention arm. BPA, best practice advisory; EPO, erythropoietin; IV, intravenous.

Daily rounding checklist for the intervention arm. This rounding checklist will be reviewed daily by the treatment team during team-based rounds for patients randomised to the intervention arm. ABG, arterial blood gas; CBC, complete blood count; GI, gastrointestinal; ICU, intensive care unit, MV, mechanical ventilation; SC, spinal cord; TBI, traumatic brain injury.
Subjects randomised to the intervention arm will receive pharmacological treatment tailored to anaemia aetiology. Patients with iron-responsive anaemias will receive a single total dose infusion (ie, 1000 mg) of low molecular weight iron dextran. Patients with anaemias of inflammation will receive a single 40 000-unit dose of subcutaneous EPO, which will be preceded by 1000 mg of iron dextran to replenish usable iron stores for those with serum ferritin levels <1000 ng/mL. Targeted pharmacological anaemia therapies are being employed to optimise efficacy and safety. For example, anaemias occurring in the presence of iron deficiency are iron-restricted and likely to respond to iron supplementation alone; hence, the potential harms of EPO likely outweigh benefits. The medical records of enrolled patients will be reviewed daily by study personnel for the duration of index hospitalisation to ensure adherence to intervention protocols (ie, use of microdraws, appropriate triggering of best practice alerts, appropriate administration of assigned study drug). Treatment may be modified or discontinued at the discretion of the clinical team if there is concern for patient safety, with all modifications recorded as study protocol violations.
Blinding
Patients randomised to control will receive standard care without an active placebo, as the multifaceted nature of the study intervention makes it difficult to design a placebo for each intervention element. As such, patients and clinicians will not be blinded to treatment assignment. Clinicians will be asked to not modify clinical cares for patients in the control arm. Transfusion practice for patients in both arms will occur in accordance with institutional transfusion guidelines (ie, red blood cells (RBCs) indicated for haemoglobin <70 g/L or <80 g/L with concern for coronary ischaemia or impaired end-organ oxygen delivery).
Outcomes
In-person assessments will occur at 1 and 3 months post-hospitalisation. Patients will undergo phlebotomy testing for assessment of haemoglobin concentrations and iron studies. The primary outcome is the mean difference in haemoglobin at 1 month. Differences in haemoglobin concentrations will also be assessed at ICU discharge, hospital discharge and 3-month post-hospitalisation. Secondary outcomes include the number and volume of phlebotomy draws throughout hospitalisation, and the number of redraws secondary to insufficient volume for laboratory assessment. Feasibility for is predefined as an incidence <5% of laboratory redraws. Patient-reported quality of life and anaemia-related fatigue will be assessed at hospital discharge, 1 and 3 months post-hospitalisation using EQ-5D (EuroQol 5 Dimension) and the 13-question Functional Assessment of Chronic Illness Therapy-Fatigue Scale, respectively. Adverse events (AEs) will be assessed through 3 months post-hospitalisation, including venous thromboembolism, bloodstream infection, myocardial infarction and stroke. Functional outcomes will be assessed at 1 and 3 months, employing the National Heart, Lung and Blood Institute (NHLBI)-funded Core Outcome Measurement Set for survivors of critical illness.14 15 This includes validated measures of physical function (6-minute walk distance, activities of daily living (ADL) survey), cognition (Montreal Cognitive Assessment Blind), mental health (Hospital Anxiety and Depression Scale (HADS), Impact of Events Scale-Revised) and the previously mentioned EQ-5D. Differences in allogeneic RBC transfusion rates will be assessed through 3 months, and a mediation analysis may be performed to determine whether the relationship between randomisation group and haemoglobin concentrations is mediated by RBC transfusions. Unplanned hospital readmissions and mortality will be assessed through 12 months. Blood samples obtained at enrolment and 1-month and 3-month follow-up (5 mL at each time point) will be stored at the study institution for biomarker assessment in future mechanistic studies on anaemia development and recovery. Compliance with treatment assignment will be reported as the proportion of subjects receiving appropriate pharmacological therapy in the treatment arm.
Data collection and handling
Data collection will be performed by trained clinical trial staff under the supervision of the principal investigator (PI, MAW). Data will be entered into electronic case report forms using REDCap. A schedule of study activities is provided in figure 3, with further details of collected data elements available in online supplemental table 2.

{kind=link}
{kind=link}
{kind=link}
Proposed study activities. AEs, adverse events; EQ5D, EuroQol 5 Dimension; EPO, erythropoietin; ICU, intensive care unit; IV, intravenous.
Sample size estimation
Power/sample size are calculated to detect a difference in haemoglobin at 1-month follow-up. Preliminary data show mean (SD) haemoglobin levels of 108 (15) g/L at 1 month among 636 patients with similar inclusion/exclusion criteria receiving standard care. A total sample size of 74 (37 per group) provides 80% power to detect 10 g/L difference using a two-sample unequal variances t-test with two-sided alpha of 0.05 to compare 1 month haemoglobin between randomised arms. Actual power is expected to be higher adjusting for pre-randomisation prognostic variables to reduce residual variation. If adjustment variables account for 25% of the variation in 1-month haemoglobin, the sample size of 37 per group provides 90% power to detect 10 g/L difference using analysis of covariance. While the analysis will employ a linear mixed-effects model (LMM), power is not appreciably different. Given expected dropout of up to 25% (death, loss to follow-up), 100 subjects will be enrolled.
Statistical considerations
As the primary approach, subjects will be analysed using an intention-to-treat approach, including all patients randomised and analysed by their assigned arm. Secondarily, analyses will be performed using modified intention-to-treat (mITT), allowing exclusion of patients who withdraw consent prior to ICU discharge as this is unlikely to be related to the assigned study arm. The longitudinal trajectory of haemoglobin will be analysed with an LMM. The primary parameter of interest is a treatment group by time interaction to estimate the effect of treatment at each follow-up time point, with 1-month haemoglobin serving as the primary outcome. The model will adjust for pre-randomisation haemoglobin, age, sex, anaemia aetiology and medical versus surgical ICU setting to reduce residual variation and improve precision.
Dropout or non-response including skipped study visit and death represent two forms of missing data. We assume dropout (unrelated to death) while in the ICU is missing completely at random; patients withdrawing consent prior to observation of haemoglobin outcome at ICU discharge will be excluded from mITT analyses. Those dropping out after ICU discharge are assumed missing at random (MAR) with missingness possibly related to adjustment covariates, arm or prior observed haemoglobin values. We anticipate up to 10% ICU mortality despite exclusion of those not expected to survive hospitalisation; such subjects will not have observed haemoglobin outcomes. We also anticipate post-ICU discharge mortality. Those with ICU mortality will have ICU discharge haemoglobin multiply imputed under the MAR assumption. Thereafter, LMMs assume additional missing data at other times are MAR. In secondary approaches to the analysis of haemoglobin, we use a worst-case imputation approach assigning a value of 0 g/L following death. If residuals are not reasonably normally distributed, a generalised linear mixed effects proportional odds model will be used or individual time points may be assessed by Wilcoxon rank-sum test without covariate adjustment.
Similar analytical approaches using proportional odds models or Wilcoxon rank-sum test will apply to the assessment of functional outcomes which are not expected to satisfy regression assumptions including normality of residuals, with additional adjustment for baseline ADLs for physical outcomes. When applicable, a cut point defining a clinically actionable adverse outcome may be defined (eg, depression as HADS-Depression ≥8). Binary outcomes will be summarised as proportion and compared by randomised arm using a χ2 test. Mortality and readmission through 1-year post-discharge will be described by a randomised arm using cumulative incidence estimates, censoring subjects at last known contact with the healthcare system when status is unknown at 1 year. Inferential analyses will use log-rank tests and Grey’s test for mortality and readmission outcomes, respectively.
We do not expect treatment to affect the heterogeneity of this intervention. However, exploratory analyses will assess potential for heterogeneity of treatment effect using interaction analyses in LMM models for the primary endpoint. An interaction term between randomised arm and each of sex, age, anaemia aetiology and surgical versus medical admissions, will be evaluated separately. The estimate of the treatment effect will be reported in subgroups using linear contrasts with the interaction analysis when evidence supports a statistically significant interaction.
In the primary analysis, the point estimate, 95% CI and p value will be reported from the LMM for the 1-month haemoglobin comparison. Since the goal is to provide robust data for further clinical trial evaluation, secondary and exploratory outcomes will be assessed without adjustment for multiplicity, with conclusions from each based on a two-sided alpha level 0.05 statistical test. There are no planned interim analyses.
Recruitment
Trained study coordinators will receive daily electronic alerts identifying ICU patients >18 years of age with a most recent haemoglobin value <100 g/L. These patients will be screened through EHR-review to ascertain eligibility. Patients, or their legal proxies in the case of a patient’s inability to directly communicate with study personnel (ie, deep sedation), will then complete a brief enrolment questionnaire to confirm no exclusions prior to written informed consent.
Retention
Given the need for in-person evaluations at 1 and 3 months post-hospitalisation, we have restricted enrolment to residents residing locally (ie, patients that receive routine medical cares in the local or regional Mayo Clinic Health System), a population with a high level of community engagement in clinical research post-hospitalisation medical cares obtained at the Mayo Clinic and its affiliated regional sites. Additional retention will be facilitated through employment of published cohort retention tools for critical care outcomes research (http://improvelto.com).16 This includes usage of a defined cohort retention protocol, collection of multiple sources of patient contact including proxies, reminder notifications, remuneration (ie, US$25 per follow-up visit) and parking vouchers.
Study funder
The study is supported through grant K23HL153310 (MAW) from the NHLBI of the US National Institutes of Health (NIH). Trial design and conduct are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Additional support for clinical coordination will be provided by the Mayo Clinic Department of Anesthesiology and Perioperative Medicine, Rochester, Minnesota, USA.
Patient and public involvement
Patients and the public were not directly involved in the design of the study, however patient care representatives including nurses, social workers and physical therapists from the study institution provided feedback on the acceptability and barriers of proposed study interventions. Study results will be available for study participants, with formal dissemination as outlined below.
Ethics and dissemination
Participant risk
There is minimal patient risk with the employment of optimised phlebotomy or decision-support. At 1-month and 3-month follow-up, patients in both arms will undergo phlebotomy testing. There is a risk of patient discomfort, which will be disclosed at enrolment. Patients will retain the right to refuse blood draws or any follow-up measures. Additionally, 5 mL of blood will be removed at enrolment and each follow-up assessment for future studies; this is not expected to have any substantial clinical consequences.
Iron and/or EPO for the treatment of anaemia do carry tangible patient risks. However, steps are being taken to minimise risk. First, we will not enrol patients at highest risk for potential harm. Second, treatments will be targeted to the underlying aetiology of anaemia, which will optimise efficacy while minimising patient-specific risk. Further, iron will be administered as a single dose of iron dextran, an approved therapy for patients with iron-deficiency anaemia that is used in current clinical practice.17 The estimated incidence of serious AEs with current intravenous iron formulations is less than 1 in 250 000.18 Nevertheless, immediate risks include allergic reactions (<1%) and non-allergic infusions reactions (eg, myalgias, arthralgias, dizziness, <1%). Administration and additional safety details are outlined in online supplemental content.
EPO will be administered as a single 40 000-unit subcutaneous injection after iron supplementation, though iron supplementation will be withheld for those with ferritin >1000 ng/mL. Short-term risks associated with this therapy include minor non-allergic adverse reactions (eg, nausea, pruritus; estimated <10%) and major adverse reactions (eg, deep venous thrombosis, uncontrolled hypertension, myocardial infarction; estimated <1%). In a recent systematic review and meta-analysis of 21 trials in critical illness, the relative risk for mortality with EPO was 0.82 (95% CI 0.71 to 0.94).19 There were no significant differences in serious AEs or venous thromboembolism. As a longer-term risk, EPO may theoretically increase the risk of tumour progression or recurrence,20–22 though evidence suggests no significant impact on adverse oncological outcomes, particularly when used in low doses and when pretreatment haemoglobin concentrations do not exceed 120 g/L.23 24
Protection
Participant privacy and confidentiality will be maintained by conducting research activities in accordance with institutional and federal guidelines. Any proposed modifications to the study protocol will be submitted to the IRB for approval. Participant information will be securely stored for internal use. Patients are free to withdraw from participation at any time on request. A formal Data Safety Monitoring Plan will be in place throughout the study in accordance with the policies of the IRB and the study funder, the NHLBI of the NIH. There are no planned interim analyses. Medical records of all study participants will be reviewed daily for AEs during the index hospitalisation. If an AE is considered serious, unanticipated and related to the study intervention, it will be reported immediately to the IRB.
Dissemination policy
This study will comply with NIH data sharing and dissemination policies. The PI and study statistician (PJS) will have access to the final trial data set. Data will be shared through the NHLBI Data Repository. Study results will be submitted by the PI for publication in peer-reviewed journals and uploaded to ClinicalTrials.gov. Study results will be accessible for study participants and the public. Ancillary studies will be encouraged. Authorship will be determined using guidelines of the International Committee of Medical Journal Editors.
Discussion
The PABST-BR trial will test the impact and feasibility of a novel multifaceted anaemia prevention and treatment strategy in critically ill patients, setting the stage for a future multicentre trial. The trial includes multiple important secondary outcomes, including those related to multidimensional post-hospitalisation functional outcomes that may be negatively impacted by anaemia (ie, physical function, cognition, mental health, quality of life). By including strategies to prevent or attenuate the severity of anaemia development (ie, optimised phlebotomy, clinical decision support) and treat anaemia with therapies directed at the underlying aetiology of anaemia (ie, iron and/or EPO) early in the course of critical illness, we hope to observe a more substantial impact on haemoglobin recovery than what would be experienced with any intervention in isolation.
There are several limitations to this study. It is an open label given challenges with the design and implementation of placebos for each unique aspect of the study intervention. If positive signals are observed, our next step will be the design of a larger placebo-controlled trial. Second, closed loop blood sampling, which is one component of optimised phlebotomy in the intervention arm, is already standard practice for arterial line draws in our adult ICU practice; hence, patients in both arms with arterial lines will receive this intervention component. Third, fixed doses administered at a single time point are being employed for both intravenous iron and EPO to minimise study complexity and the potential for error. Future studies may be needed to optimise dosing regimens in critical illness. Finally, this pilot trial is relatively limited in size. However, the sample size has been selected after power analysis based on pre-existing patient data and is sufficient for the primary outcome of haemoglobin recovery. The trial is not powered for multiple secondary outcomes, which should be considered hypothesis-generating for a larger trial.
In conclusion, the PABST-BR trial is designed to determine if a multifaceted anaemia management strategy is feasible and associated with improved haemoglobin recovery when compared with standard care. The trial will inform a large multicentre trial to optimise haemoglobin recovery and post-hospitalisation outcomes in critical illness survivors.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MAW, PS, OG and DJK developed the trial concept and drafted the protocol manuscript. RSG, WBB, JEC, BK, AC, LM, KAD, MLJ and BKA helped to develop the trial concept and revised the manuscript critically for important intellectual content. All listed authors approved the final version of the manuscript for publication and agreed to be accountable for all aspects of the work.
Funding This work is supported by the National Heart Lung and Blood Institute (NHLBI) of the US National Institutes of Health (NIH) grant K23HL153310 (MAW). Trial design and conduct are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Additional support for clinical coordination is provided by the Mayo Clinic Department of Anesthesiology and Perioperative Medicine, Rochester, Minnesota, USA.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.