Article Text

Abstract
Introduction Endovascular treatment is the standard of care for acute large vessel occlusion (LVO) in the anterior circulation. However, successful complete recanalisation is considerably difficult when the vessels are severely tortuous. At the bend, the stent retriever can distort, collapse and lose its ability to capture the clot due to structural change. The aim of the present study is to evaluate the safety and efficacy of the new thrombectomy device multisegment Mechanical Thrombectomy (MT) System for endovascular treatment of acute ischaemic stroke (AIS).
Methods and analysis The present study is a prospective, multicentre, randomised controlled trial conducted in 11 stroke centres in China. The safety and efficacy of vascular recanalisation in patients with AIS who will be treated with a new thrombectomy device-multi-segment MT System or with Solitare FR within 8 hours of symptom onset will be compared. A total of 238 subjects who met the inclusion and exclusion criteria will be randomised into either a treatment group or a control group by an internet-based Central Random System in a 1:1 manner, and 30 subjects will be recruited into the small sample study. SAS V.9.4 statistical software will be used for statistical analysis of the primary endpoint indicators and other indicators.
Ethics and dissemination The study involving human participants was reviewed and approved by the Ethics Committee of Drugs (devices) Clinical Experiment in Henan Provincial People’s Hospital (reference number: AF/SC-07/04.0) and other research centres participating in the clinical trial. The results yielded from this study will be presented at international conferences and sent to a peer-review journal to be considered for publication. The Standard Protocol Items: Recommendations for Interventional Trials checklist was utilised when drafting the study protocol.
Trial registration number Registry on 10 September 2021 with Chinese clinical trial registry: ChiCTR2100051048.
- stroke
- neurology
- neuroradiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The small sample study will be the first trial to evaluate the efficacy and safety of the thrombectomy device for subjects with symptoms onset between 8 and 24 hours.
If the trial is successful, it will be the first third-generation thrombectomy device approved in China, and also will considerably reduce treatment costs of stroke caused by large vessel occlusion.
Subjects will be recruited from multiple stroke centres in China.
Since this trial will be conducted in mainland China, where the large artery atherosclerosis stroke subtype is more prevalent among subjects, it may limit generalisability of the study result.
The study will not be blinded to surgeons and nurses, and lacks a uniform standard for the use of adjunct devices among centres.
Introduction
Stroke is the leading cause of morbidity and mortality worldwide, among which ischaemic stroke is the more common type and patients with it account for 60%–80% of patients with stroke.1 The treatment of acute ischaemic stroke (AIS) mainly includes thrombolysis and mechanical thrombectomy (MT) in order to recanalise the occluded vessels as soon as possible and salvage the ischaemic penumbra. Recombinant tissue plasminogen activator is a common treatment used for ischaemic stroke. The time window for thrombolysis in AIS can be extended to 9 hours by a mismatch between diffusion-weighted imaging (DWI) and fluid attenuated inversion recovery (FLAIR) or automated perfusion imaging. but more than half of the treated patients do not recover completely, or die. With the continuous development of medical technology and interventional materials, endovascular treatment can significantly improve the recanalisation rate of occluded vessels, extend the time window and reduce the haemorrhage transformation rate.2–5
Several randomised controlled trials have shown that the safety and efficacy of endovascular recanalisation therapies by stent retriever over standard medical care in patients with AIS have caused occlusion of vessels of the proximal anterior circulation.6–10 The Food and Drug Administration of USA approved Solitaire (Medtronic/ev3) and (Stryker) stent retrievers to be mainly used for the treatment of large vessel occlusive (LVO) stroke in 2012. A stent retriever has the advantages of good navigation and rapid recanalisation with a lower risk of long-term complications and uses a temporary stent to capture the thrombus and restore the blood flow by removing the thrombus and directing it to the peripheral vascular wall. During stent withdrawal, the thrombus is captured into the stent space and removed together with the stent.
However, successful complete recanalisation is considerably difficult when the vessels are severely tortuous. Within tortuous vessels, the stent may distort, collapse and lose its ability to capture the clot due to structural change.11 12 The MT System, designed by NeuroVasc Technologies Inc, includes a multisegment design that allows it to remain stretched even when it is pulled and twisted, so as to attach firmly to the clot as it turns through the tortuous vessels. In addition, the distal end of the multisegment stent remains open during the removal of the clot along with the stent of the body, which is conducive in preventing the broken thrombus from escaping to the distal end.
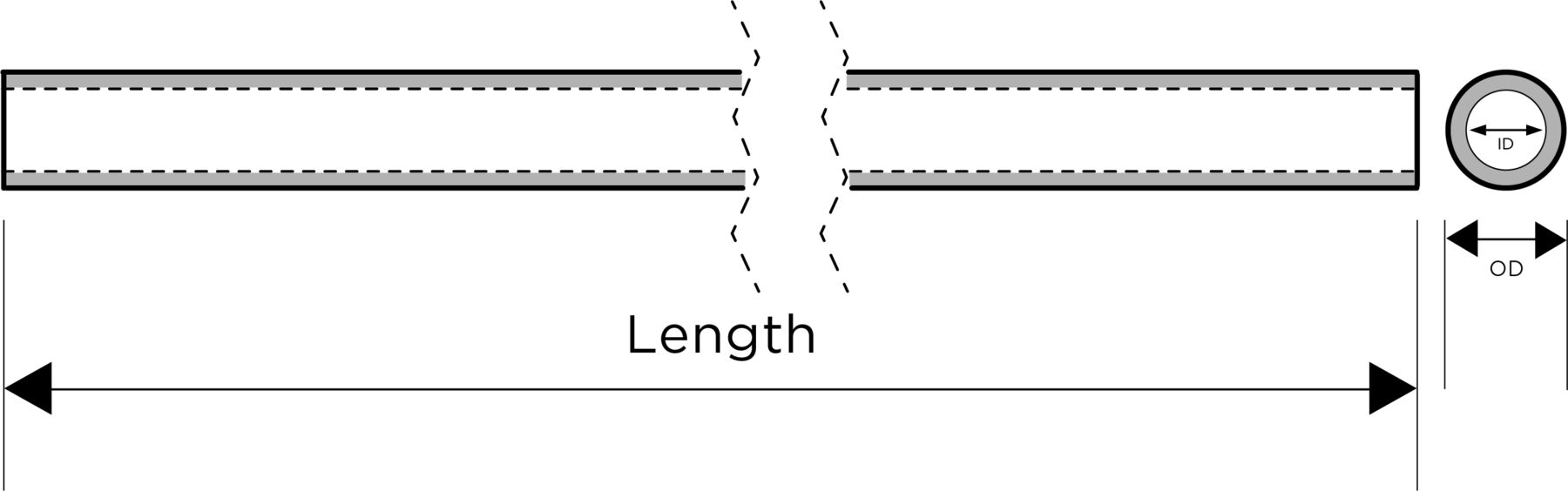
The multisegment MT System consists of a stent (figure 1), a push rod (figure 2) and an introducer sheath (figure 3). The material of the stent is a nickel–titanium alloy and the material of the label is a platinum–iridium alloy. The pushrod is made of nickel–titanium alloy, and the distal end of the pushrod is mechanically connected with the proximal end. The introducer sheath has a three-layer structure, including an inner layer, a middle layer and an outer layer. The inner layer and outer layers have different functions, while the middle layer is a tie layer that allows the inner and outer layers to bond together. The inner layer needs to provide a low friction surface for interacting with the stent retriever. The outer layer provides structural integrity for the introducer sheath to resist compromising the lumen in normal handling or when compressed by a rotating haemostasis valve. The stent is preinstalled in the introducer sheath. The inner diameter of the compatible micro catheter is 0.165/0.021. According to the stent diameter, it is divided into different specifications of 3.5 mm, 5 mm and 6 mm.

The diagram of the stent.

The diagram of the push rod.
{kind=link}
{kind=link}
{kind=link}
The diagram of guiding the sheath. ID, inner diameter; OD, outer diameter.
The purpose of the present study is to evaluate, in a prospective, randomised, controlled trial the safety and efficacy of the multisegment MT System conducted by NeuroVasc Technologies Inc for patients with AIS and with anterior circulation LVO, and provide a theoretical basis for the official listing and application of the multi-segment MT System.
Methods
Design
This trial consists of a prospective, multicentre, randomised controlled, non-inferiority study and a small sample study. The aim is to evaluate the safety and efficacy of the multisegment MT System conducted by NeuroVasc Technologies Inc for endovascular treatment of patients with AIS due to LVO within 8 hours of disease onset. The planned start date for this study was November 2020, and planned end date is November 2022. The subjects will be recruited in 11 high-volume comprehensive stroke centres in China, and the study protocol has been approved by the Ethics Committee of Drugs (Devices) Clinical Experiment in Henan Provincial People’s Hospital and other research centres participating in the clinical trial. A total of 238 subjects who meet the inclusion and exclusion criteria will be randomised into either a treatment group or a control group by a central web-based system in a 1:1 manner, and 30 subjects with symptoms between 8 and 24 hours will be recruited into the small sample study. The treatment group will receive MT with the suitable size of the multisegment MT System (NeuroVasc Technologies Inc), and the control group will be treated with Solitare FR. The patients with 8–24 hours of symptom onset will be enrolled in the small study. Table 1 is a brief summary of the visits and the assessment schedule.
Brief summary regarding the visits and the assessment schedule
Participants
Detailed study inclusion and exclusion criteria are shown in table 2. Treatment allocation occurs when the study participant meets all of the inclusion criteria and signs the informed consent form.
Detailed study inclusion and exclusion criteria
Treatment and intervention
Subjects who are eligible for intravenous thrombolysis will be administered 0.9 mg/kg (maximum dose 90 mg) Alteplase according to AHA/ASA, while endovascular treatment is prepared. For all the eligible subjects, a stent retriever should be provided as the first-line approach. Multisegment MT System will be used for the study group and the Solitare FR for the control group. An interventional neurologist will select the intravenous sedation or general anaesthesia based on the subject’s clinical condition to ensure the subject’s comfort and safety. DSA will be performed to determine the location of the occluded vessels, vascular path and collateral circulation compensation. Stent retriever thrombectomy can be performed three times or less before rescue therapy. If the intracranial occluded blood vessel had significant stenosis or other lesions after MT with stent retriever, the researcher should evaluate whether to perform the aspiration thrombectomy, balloon dilation, stent implantation or other rescue therapy based on the subject’s clinical conditions. extended Thrombolysis In Cerebral Infarction (eTICI) classification will be used to assess and evaluate the condition of recanalisation of the occluded vessels, and the Dyna-CT scan will be performed immediately after the operation to observe whether intracranial haemorrhage has occurred.
Randomizsation
All 238 subjects are randomly allocated into the study group or the control group at a 1:1 ratio based on an internet-based Central Random System. Randomisation will be performed after the completion of brain DSA immediately prior to MT. In order to control for key indicators during grouping, the clinical trial centre and baseline NIHSS scores (NIHSS score <16 and NIHSS score ≥16) were used as stratification factors.
Reduction and avoidance of bias
The trial will be conducted in multiple stroke centres, and the case samples from multiple centres will be more representative than that from a single-centre, preventing the systematic error of a single-centre from leading to the bias of the trial results and resulting in more reliable conclusions. At the same time, in order to reduce operational differences among centres, the study-related personnel in each centre will be trained in a unified standard, and the evaluation criteria for efficacy and safety indicators will be adopted. An independent third-party core laboratory will be used to evaluate the endpoint of successful vascular recanalisation rate (eTICI classification).
Monitoring plan
Prior to the initiation of the clinical trial, the sponsor/agent, the supervisor and the head of each centre should train the investigator on the study protocol so that the investigator can understand and be familiar with the trial product. The establishment of an inspection plan will be performed as follows: qualified inspectors will be appointed by the sponsor or the agent to conduct regular on-site inspection visits to each centre and ensure that all contents of the study plan are strictly followed. Monitoring will be performed by the e-CRF to ensure consistency with the content of the original data.
An endpoint Event Committee (CEC) will be established in the present study to conduct a blind review and to evaluate the endpoint events occurring during the trial. The Endpoint Event Committee (CEC) is composed of clinical experts who are independent of the investigator and the sponsor and are not directly involved in the implementation of the clinical trial. These clinical experts are in the relevant clinical research area. No interim analysis will be performed in this study.
Sample size
The inclusion criteria of the selected subjects were strictly limited in this protocol. All the selected subjects belonged to the same disease and were further divided according to the time of symptom onset as follows: assuming that the rate of successful recanalisation of subjects in the control group after treatment is approximately 88% based on the available clinical evidence and the experience of neurointerventionists,8 and it is expected that the study group can achieve the same efficacy. A total of 238 patients are expected to be enrolled in the randomised controlled study within 8 hours of symptom onset, 119 cases in each group with a 12.5% non-inferiority margin, a 5% significance level (two-tailed), an 80% statistical power and a 10% dropout rate. Due to no thrombectomy devices suitable for such indications, the efficacy and safety of the multisegment MT System for subjects with symptoms between 8 and 24 hours cannot be verified in a randomised controlled manner. So a small sample of the subjects with symptoms between 8 and 24 hours will be included in the study with an expected enrolment of 30 patients who all treated with the multi-segment MT System.
The present study will be carried out in multiple stroke centres at the same time. In principle, the number of enrolled centres will be evenly distributed as far as possible to ensure adequate centre representation. However, considering the feasibility and inclusion progress, the number of subjects will be adjusted according to the actual situation to ensure the balance of the inclusion scale of each centre. The final inclusion scale of a specific centre should not exceed 50% of the total number of cases.
Patient and public involvement
There was no patient and public involvement in this protocol.
Results
Primary efficacy endpoint
The primary efficacy endpoint of the study is the successful recanalisation rate. The Core Lab will assess whether the target vessels have achieved successful recanalisation by DSA, which is defined as eTICI 2b or greater revascularisation. The successful recanalisation rate will be estimated as follows: Number of successful arterial recanalisation/number of targets of stent thrombectomy.
Secondary efficacy endpoints
The study has the following eight secondary clinical efficacy endpoints: (1) time of arterial recanalisation defined as the time from groin puncture to eTICI 2b or better revascularisation; (2) NIHSS scores, which will be estimated at preoperation, 24 ± 6 hours postoperation and 7 ± 2 days postoperation or before discharge to evaluate the neurological condition; (3) ratio of mRS 0–2 at 90 ± 14 days postoperation, which is used to evaluate the neurological condition and optimal neurological outcome defined as mRS <2; (4) rate of device success, which indicates that the thrombectomy device was successfully deployed through the occlusive vessels and that the delivery system was successfully recycled; (5) first-pass successful/near-perfect reperfusion (defined as eTICI ≥2b); (6) rate of surgical success, which is calculated by the proportion of successful surgical cases to all subjects on the basis of eTICI 2b or greater revascularisation; (7) rate of distal vascular embolism, which indicates a new embolisation of the distal end of the occlusive vessels; (8) time of MT.
Safety outcomes
The present study exhibits the following five safety outcomes: (1) rate of symptomatic intracranial hemorrhage (sICH) at 24 ± 6 hours, which is defined as any ICH event associated with neurological deterioration of an increase of 4 points or more on the NIHSS score compared with that of the preoperative state; (2) rate of asymptomatic intracranial hemorrhage (aICH) at 24 ± 6 hours, which is defined as any intracranial hemorrhage (ICH) event associated with neurological deterioration corresponding to an increase of under 4 points on the NIHSS compared with the preoperative state; (3) rate of severe adverse events related with the device or surgery at 7 ± 2 days postoperation or before discharge; (4) rate of mortality related with surgery at 7 ± 2 days following the operation or before discharge; and (5) rate of all-cause mortality at 90±14 days following the operation.
Statistical analysis
For descriptive analysis, the enumeration data will be described by frequency and component ratio, and the measurement data will be described by mean, SD, maximum, minimum, and median, as well as 25th and 75th quantiles.
For baseline demographic analysis, the likelihood ratio χ2 test will be used for comparison of the enumeration data between the groups based on the descriptive analysis, and the Fisher’s exact test will be used when more than 25% of the theoretical frequency is <5; the normally distributed measured data between the groups will be compared by the group t-test, and the non-normally distributed measured data between the groups will be compared by the Wilcoxon rank-um test.
For primary efficacy endpoint of successful arterial recanalisation, the comparison between the groups will be performed using the Cochran-Mantel Haenszel (CMH) χ2 analysis of the adjusted centre effect. In addition to the success rate of the study and the control groups, the difference in the success rate between the groups and the 95% CI will also be estimated. The method of other efficacy indicators for between-group comparisons should be the same as that of the baseline analysis. The intragroup comparison of the normally distributed data will be performed by the paired t-test and the Wilcoxon signed-rank test will be used for the intragroup comparison of non-normally distributed data.
For evaluation of safety outcomes, the number and proportion of cases that are normal before treatment and abnormal after treatment will be described. The number and incidence of adverse events will be described, and the proportion will be tested by the likelihood ratio χ2 test and Fisher’s exact test. Moreover, the specific manifestations and extent of all adverse events in each group and their relationship with the study devices will be described in detail.
For the primary endpoint indicators, statistical analysis will be performed at a unilateral significance level of 0.025 (corresponding to the unilateral confidence limit of 95% CI), and the statistical analysis of the other indicators will be performed at a significance level of 0.05. SAS V.9.4 statistical software will be used for statistical analysis.
Ethics and dissemination
The protocol of this study has been approved by the the Ethics Committee of Drugs (devices) Clinical Experiment in Henan Provincial People’ s Hospital (reference number: AF/SC-07/04.0) and other research centres participating in the clinical trial. The study has been registered at Chinese Clinical Trial Registry (registration number: ChiCTR2100051048), and this protocol is accordance with the Declaration of Helsinki and relevant laws and regulations in China. All patients or their legal representatives will have to sign the written informed consent (online supplemental file 1). It is imperative that participants have the right to withdraw from research at any time. Any safety issues related to study, such as modifications of the study protocol or informed consent form of the subjects and serious adverse events during the clinical trial, must be reported to the Ethics Committee in time.
Supplemental material
The database used during the study will be available from the corresponding author on reasonable request. The results of the trial will be disseminated through a series of peer-reviewed publications and conference presentations. We used the Standard Protocol Items: Recommendations for Interventional Trials checklist when writing our study protocol.
Discussion
The development of the new MT devices has enabled the widespread use of the stent retriever for stroke caused by LVO and the significant improvement in the rate of successful recanalisation.13 However, certain side effects can occur including the escape of the thrombus to the distal end, and the deformation of the stent or its distortion at the bend during the clot. The purpose of the present trial was to evaluate the safety and efficacy of the multisegment MT System conducted by NeuroVasc Technologies Inc. compared with the Solitare FR in patients diagnosed with AIS.
The multisegment MT System exhibits certain unique characteristics compared with the previously used stent retriever.14–16 The multisegment design of the multisegment MT System keeps the stent open in the diameter direction under tension, thus improving the ability to capture and remove the clot. Similarly, the multisegment MT System has a stronger grasping ability for the clot compared with other stent retriever systems.17 In addition, inlaid marks on each stage and throughout the working length provide X-ray visibility of the stent, which can aid the doctor to determine the length and position of the stent and its unfolding shape. This leads to the improvement of the rate of successful recanalisation of the target vessels. Due to the gradual flexibility, the stent can pass through the twists and turns of the internal carotid artery (ICA) and the middle cerebral artery during delivery, such as the cavernous segment of the ICA. The multisegment MT System has different sizes and specifications, which can meet the requirements of the occluded vessels with different diameters. Compared with first-generation devices, Solitare FR has been confirmed to achieve superior recanalisation rates, faster reperfusion, lower haemorrhagic transformation complications and improved clinical outcomes.18 Therefore, it is reasonable to select Solitare FR as a control product in this trial.
There are some limitations in the study design. First, since this trial will be conducted in mainland China, where the large artery atherosclerosis stroke subtype is more prevalent among subjects, it may limit generalisability of the study result. Second, the study will not be blinded to surgeons and nurses, and lacks a uniform standard for the use of adjunct devices among centres.
In conclusion, the results of this trial will provide information on the safety and efficacy of the multi-segment MT System. The success of this trial will provide a new MT device for the treatment of AIS in China.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
HZ and ZL are joint first authors.
Contributors TL and YH conceived the study. ZL designed the study. HZ contributed to the draft of the manuscript. ZL, TZ and YH contributed to the revision of the manuscript. All authors read and approved the final manuscript.
Funding The present trial was funded by the National Stroke High-risk Population Intervention Technology Research and Promotion Project (GN-2018R0007) Provincial and Ministerial Joint Project of Henan Provincial Medical Science and Technology (SBGJ202002001). The present trial was supported by NeuroVasc Technologies Inc. (no grant number), and the company provided the stent retrievers for free.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.