Article Text

Abstract
Introduction The purpose of this study is to assess the ability of two new ECG markers (Regional Repolarisation Instability Index (R2I2) and Peak Electrical Restitution Slope) to predict sudden cardiac death (SCD) or ventricular arrhythmia (VA) events in patients with ischaemic cardiomyopathy undergoing implantation of an implantable cardioverter defibrillator for primary prevention indication.
Methods and analysis Multicentre Investigation of Novel Electrocardiogram Risk markers in Ventricular Arrhythmia prediction is a prospective, open label, single blinded, multicentre observational study to establish the efficacy of two ECG biomarkers in predicting VA risk. 440 participants with ischaemic cardiomyopathy undergoing routine first time implantable cardioverter-defibrillator (ICD) implantation for primary prevention indication are currently being recruited. An electrophysiological (EP) study is performed using a non-invasive programmed electrical stimulation protocol via the implanted device. All participants will undergo the EP study hence no randomisation is required. Participants will be followed up over a minimum of 18 months and up to 3 years. The first patient was recruited in August 2016 and the study will be completed at the final participant follow-up visit. The primary endpoint is ventricular fibrillation or sustained ventricular tachycardia >200 beats/min as recorded by the ICD. The secondary endpoint is SCD. Analysis of the ECG data obtained during the EP study will be performed by the core lab where blinding of patient health status and endpoints will be maintained.
Ethics and dissemination Ethical approval has been granted by Research Ethics Committees Northern Ireland (reference no. 16/NI/0069). The results will inform the design of a definitive Randomised Controlled Trial (RCT). Dissemination will include peer reviewed journal articles reporting the qualitative and quantitative results, as well as presentations at conferences and lay summaries.
Trial registration number NCT03022487.
- ischaemic heart disease
- pacing & electrophysiology
- heart failure
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study will provide strong validation of the performance of Regional Repolarisation Instability Index (R2I2) and Peak Electrical Restitution Slope (PERS) as predictors of sudden cardiac death in a cohort of patients with ischaemic heart disease.
R2I2 and PERS have previously shown strong positive predictive values suggesting that they may be of use in lower risk populations.
This study is investigating a cohort of patients undergoing implantable cardioverter-defibrillator (ICD) implant; as such the primary endpoint of ventricular fibrillation or sustained ventricular tachycardia >200 beats/min is an imperfect surrogate for sudden cardiac death.
A positive outcome would lead to a future study using R2I2/PERS to determine ICD implantation with all cause mortality as the primary endpoint.
Introduction
Sudden cardiac death (SCD) is a major cause of mortality accounting for 4–5 million deaths per year worldwide with coronary heart disease being the underlying aetiology in 80% of cases.1 Implantable cardioverter-defibrillator (ICD) technology has developed rapidly over the last four decades with the indications for implantation broadening in light of new evidence. Large randomised controlled trials have established their use in primary indications for preventing SCD and improving overall survival provided that patients are appropriately selected.2–6 Implant rates continue to rise with 238 per million population high energy devices implanted in England between 2015 and 2016.7 Despite this, SCD remains an important cause of mortality because of the limitations of current risk assessment. The majority of deaths occur in those considered to be low risk using current stratification criteria.8 Also, a substantial proportion of ICD recipients do not require therapy from their devices and ICDs carry a substantial morbidity with possible complications such as infection and inappropriate shocks.9 Imprecise ICD prescription can result in unnecessary cost with often suboptimal selection based on crude clinical parameters. SCD risk stratification therefore remains inadequate and a priority area for clinical research.
Action potential duration (APD) restitution describes an inherent property of the myocardium whereby the APD is dependent on the preceding diastolic interval (DI). The DI is the rest period between repolarisation and the next excitation. The relationship between APD and DI can be plotted on a restitution curve.10 The APD lengthens with longer DIs but at shorter DIs the APD restitution curve steepens such that small changes in DI cause a large change in APD.
APD restitution has been shown to be an important property in the genesis and maintenance of ventricular arrhythmia (VA). Ventricular fibrillation (VF) consists of multiple spiral re-entrant waves which have short lifetimes and require continual generation in order to persist and propagate. This is provided by wavebreak with electrical wavefront splitting into multiple wavelets.11
APD restitution has been proposed to be associated with arrhythmogenesis by two different mechanisms. First, the ‘restitution hypothesis’ states that the main determinant of wavebreak is the steepness of the restitution curve. When the gradient of the restitution curve is >1, small changes in DI can lead to a large change in APD and oscillations between APD and DI are magnified. Computer and experimental models have shown that steep curves promote instability and spiral wave breakup which in turn can lead to VF.12 13 Second, heterogeneity of APD restitution in adjacent myocardium allows wavefronts to become dissonant, thereby providing a substrate for wavebreak and re-entry. APD restitution properties within the ventricle have been shown to display interventricular, intraventricular and transmural heterogeneity.14–16
Two novel surface ECG markers have been developed based on the principles of these mechanisms.17 The Regional Restitution Instability Index (R2I2) reflects heterogeneous restitution behaviour in different regions of the heart quantified using interlead heterogeneity. It is derived from the difference of the mean SD of the residuals from the mean gradient for each ECG lead across the range of DIs. In an initial retrospective study in patients with ischaemic cardiomyopathy, R2I2 was shown to be associated with VA or death.18 It was subsequently evaluated in a prospective, blinded study of 60 patients where it successfully replicated the findings of the retrospective study. R2I2 was found to be significantly higher in those reaching the endpoint of VA/SCD compared with those that had not (1.11±0.09 vs 0.84±0.04, p=0.003). Using a predefined cut-off value from the retrospective study, patients with R2I2>1.03 had significantly higher rates of VA/SCD (p<0.0001) with a sensitivity, specificity, positive predictive value and negative predictive value of 63%, 82%, 56% and 86%, respectively.19
A second surface ECG marker that measures peak APD restitution gradient was also assessed within the same cohort of patients with ischaemic cardiomyopathy.19 Peak ECG Restitution Slope (PERS) was calculated as the mean of the peak restitution slopes across the 12 ECG leads. PERS was significantly higher in those experiencing VA/SCD than those that did not. Patients with PERS >1.21 were shown to have an incidence ratio of 4.1 times than those with PERS <1.21. Combining both biomarkers, a R2I2>1.03 and PERS >1.21 gave a sensitivity, specificity, positive predictive value and negative predictive value of 50%, 95%, 80% and 84%. The relative risk of VA/SCD was 21.6 when compared with R2I2<1.03 and PERS<1.21 combined. The predictive utility of both markers for VA/SCD was independent of left ventricular ejection fraction (LVEF) and QRS duration. The two parameters were also independent of each other supporting the view that they reflect different arrhythmogenic mechanisms.
Although the findings from this study were statistically significant and replicated the findings of the initial retrospective study, a relatively small number of participants were recruited from a single UK centre. ICDs were implanted for both primary and secondary indications in the prospective study with devices implanted for a secondary prevention indication accounting for the greatest proportion of VA/SCD endpoints. This potentially questions whether these ECG markers would retain significant predictive value in the primary prevention population given that implanting a device would need greater justification in such a patient. Both VA and SCD were combined as a single endpoint. Given that VA may be asymptomatic, self-terminating and not resulting in SCD, this combined endpoint would not adequately reflect the predictive risk of mortality.
The Multicentre Investigation of Novel Electrocardiogram Risk markers in Ventricular Arrhythmia prediction (MINERVA) study has been designed to address these issues aiming to further stratify VA and SCD risk for primary prevention in a multicentre trial. The MINERVA Investigators group is part of the British Heart Rhythm Society Multicentre Trial Collaboration.
Study design
MINERVA is a prospective, open label, single-blinded, multicentre observational study. The study is registered with ClinicalTrials.gov (NCT03022487) and National Institute of Health Research in the UK (trial no. 31324). Patients with ischaemic cardiomyopathy undergoing a first time ICD or Cardiac Resynchronisation Therapy-Defibrillator (CRT-D) implantation for primary prevention (according to UK NICE guidelines20) will undergo a standard 30 min electrophysiological (EP) cardiac stimulation protocol performed at the end of the implant procedure. The EP test will be performed while recording a high-resolution digital 12-lead ECG. There is no randomisation required as all study participants will receive EP study at baseline. Standard clinical follow-up will take place through the ICD clinic as per local arrangements. Blinding will be maintained at the core lab where the ECG analysis will be performed. The core lab will not have details of patient health status. The results derived from the EP study data will be correlated with event rates to establish the relationship of the ECG markers in predicting VA risk. The study is event-driven with a sample size of 440 patients. Recruitment for this study has begun with the first patient having been recruited in August 2016. The minimum follow-up period should be 18 months and a maximum of 3 years.
Patient and public involvement
The science underpinning this study and this research study were presented to patients and the public 17 November 2015.
Objectives
The primary objective is to investigate whether R2I2 and PERS are significantly higher in ischaemic cardiomyopathy patients reaching the endpoint of VA and/or SCD than those who do not during the follow-up period. The secondary objectives are to assess whether significantly more endpoints are reached in patients with any of the following:
R2I2>1.03 in comparison to R2I2<1.03.
PERS>1.21 in comparison to PERS<1.21.
Both R2I2>1.03 and PERS >1.21 in comparison to both R2I2<1.03 and PERS<1.21.
The primary endpoint is VF or sustained ventricular tachycardia at a rate greater than 200 beats/min as recorded by the ICD. The secondary endpoint is SCD. The endpoints will be adjudicated by the Endpoint validation committee.
Patients
The intended population for this study are patients with ischaemic cardiomyopathy attending for a de novo ICD implantation (including CRT-D) for primary prevention indications based on current National Institute for Health and Care Excellence (NICE) guidelines (table 1).20 Patients at 15 UK centres (online supplemental appendix 1) who meet the inclusion and exclusion criteria, as detailed below, will be considered for the study.
Supplemental material
Treatment options with implantable cardioverter-defibrillator (ICD) or CRT for people with heart failure who have left ventricular dysfunction with an Left ventricular ejection fraction of 35% or less (according to New York Heart Association (NYHA) class, QRS duration, LBBB, left bundle branch block)
Inclusion criteria
To be included in the study, patients must be aged over 18 years of age, able to give informed consent for participation, and able to comply with the study requirements. Female patients of childbearing potential must be willing to ensure the use of effective contraception by themselves or their partner during the course of the study. All patients must have a diagnosis of ischaemic cardiomyopathy, on stable medication (as defined as no more than a 100% increase or 50% decrease in current regular medication for at least 4 weeks prior to study entry), and be attending for a de novo, primary prevention ICD implantation based on NICE technology appraisal (TA314). They must be able to read and understand English and allow their general practitioner (GP) and consultant, if appropriate, to be notified of participation in the study. Lastly, they must be able to and agree to attend follow-up at the study site until the closure of the study.
Exclusion criteria
Patients are excluded from this study if they are within 28 days of an acute coronary syndrome or cardiac surgery, scheduled for elective surgery or any procedure requiring general anaesthesia, are pregnant, lactating or planning a pregnancy during the course of the study, have significant renal disease (requiring renal replacement therapy and/or estimated Glomerular Filtration Rate (eGFR)<15), severe liver disease (end stage or the presence of liver cirrhosis), are participating in another research study involving an investigational product in the last 12 weeks, are undergoing any interventional research, have contraindications for an EP study including haemodynamic instability, severe valvular pathology as defined by the British Society of Echocardiography guidelines, have symptomatic coronary artery disease, or had a stroke within the last 12 months. Participants will be excluded if they have a significant disease or disorder which, in the opinion of the investigator, may put the participant at risk because of participation in the study, or may influence the result of the study, or affect the participant’s ability to participate in the study. At the time of ICD implantation the following exclusions from the study apply: the right ventricular lead is not apically positioned, if it is adjudged by the implanting physician that the patient would require a ventricular tachycardia (VT) therapy zone ≤200 beats/min or if there is any ventricular bradycardia pacing indication.
Patient randomisation
No randomisation is required as all study participants will receive the EP study at baseline.
Study plan
The pathway for the study is shown in figure 1. The minimum follow-up period is 18 months. The study opened 8 July 2016 and will close 30 June 2021, patients have been recruited by 15 different centres in the UK. Recruitment has been substantially prolonged because of the COVID-19 epidemic.
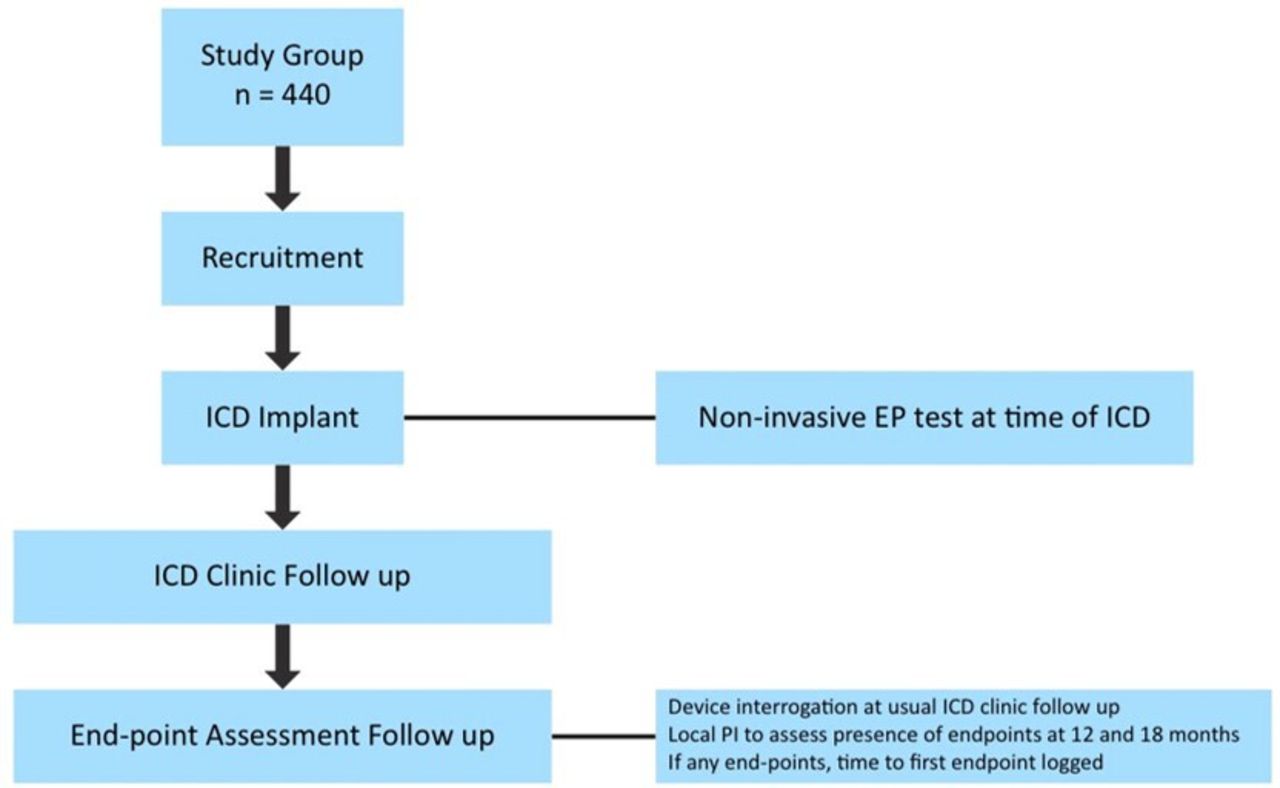
Study flow chart. EP, electrophysiological; ICD, implantable cardioverter-defibrillator; PI, Principal Investigator.
Screening and eligibility assessment
Only patients already destined for implantation of an ICD will be approached to consider participation in the study. They may be identified from either an inpatient or outpatient referral process. Once identified, the research team at each site will confirm the suitability of individual patients by reviewing their medical history and notes. Eligibility will be confirmed from the assessment of basic demographics, medical history, concomitant medication, recent ECGs, cardiac function and blood tests. After informing the care team, the research team will then approach eligible patients either in person, when they are visiting the hospital for routine outpatient appointments, while they are inpatients awaiting the procedure itself, or via telephone conversation to enquire if they would like to be considered for the study. The patient information leaflet and informed consent form will be given or sent to the patient for full consideration. They will be allowed a minimum of 24 hours to consider the information. Written informed consent will be obtained either at a subsequent visit or on the day of the implantation of the ICD.
Baseline assessment
Following recruitment, demographic information, medical history, concomitant medication, basic blood chemistry, ECG parameters and LVEF (as assessed by either echocardiography or MRI) will be confirmed and documented as baseline data.
Non-invasive EP study via ICD
At ICD implantation, the deliverable therapy zones will be programmed at rates >200 beats/min with a monitor-only zone from 150 beats/min. The parameters will otherwise be programmed according to the manufacturer specific guidelines as per the 2015 Consensus Statement on Optimal ICD Programming and Testing.21
The EP study is performed using a single extrastimulus protocol as per the standard non-invasive physiological stimulation function of ICDs from all manufacturers and will be performed through the device immediately post implantation.
Recording of the digital 12-lead ECG during the EP study protocol will be performed using a portable high-resolution 1 kHz sampling digital ECG recorder (Norav 1200-HR (Norav Medical, Weisbaden, Germany)) and the company software and hardware (Universal Serial Bus (USB) dongle) included with the Personal Computer (PC) interface. The risks of the non-invasive EP study are small (1.3% risk of arrhythmia).22 Unlike a standard VT stimulation study, the aim is not to provoke VA. The objective is simply to obtain a range of values from which to derive R2I2 and PERS. The non-invasive EP procedure would add no more than 30 min to the standard care of the ICD implant.
Programmed stimulation will be delivered at the right ventricular (RV) apex via the ICD using the manufacturer-specific programmer. Rectangular pulses will be delivered with a pulse duration of 2 ms and output at three times the diastolic threshold. The drive train (S1) length is 10 beats followed by one extrastimulus (S2). For valid data, the final two S1 of the drive train and S2 must successfully capture in succession (figure 2), or else the drive train would be repeated.

Example of captured stimulus. For valid data, the final two S1 of the drive train and S2 must successfully capture in succession, or else the drive train should be repeated.
The EP study protocol consists of two stages which are both repeated. For stage 1, a drive cycle length (DCL) of 600 ms is followed by a single S2 at 500 ms. Drive trains are to be repeated with S1−S2 coupling interval decremented by 20–300 ms and then by 10 ms to the effective refractory period (ERP). If breakthrough beats are seen consistently in the drive train, the DCL should be reduced to 500 ms with the S1−S2 interval starting at 460 ms.
For stage 2, the DCL is 400 ms with the initial coupling interval at 360 ms, decremented by 20–300 ms and then by 10 ms to ERP.
Subsequent assessment and follow-up
Standard clinical follow-up will take place through the ICD clinic as per local arrangements. The initial appointment usually takes place 4–6 weeks post implantation and subsequently every 6 months. During the appointments, an ICD interrogation will be performed with the report exported as part of routine clinical care by a cardiac physiologist or a suitably trained investigator. Some centres may use home monitoring. During the appointments, the research team will recheck eligibility, reconfirm willingness to participate, assess and record the presence of arrhythmia-related endpoints, record current medications, and report any adverse events (AE). Information not requiring device interrogation can be obtained through telephone interviews. At preset time intervals (12 and 18 months), the local PI will assess the presence of endpoints from patient notes and record the exact time to the first endpoint if present.
Analysis of R2I2 and PERS
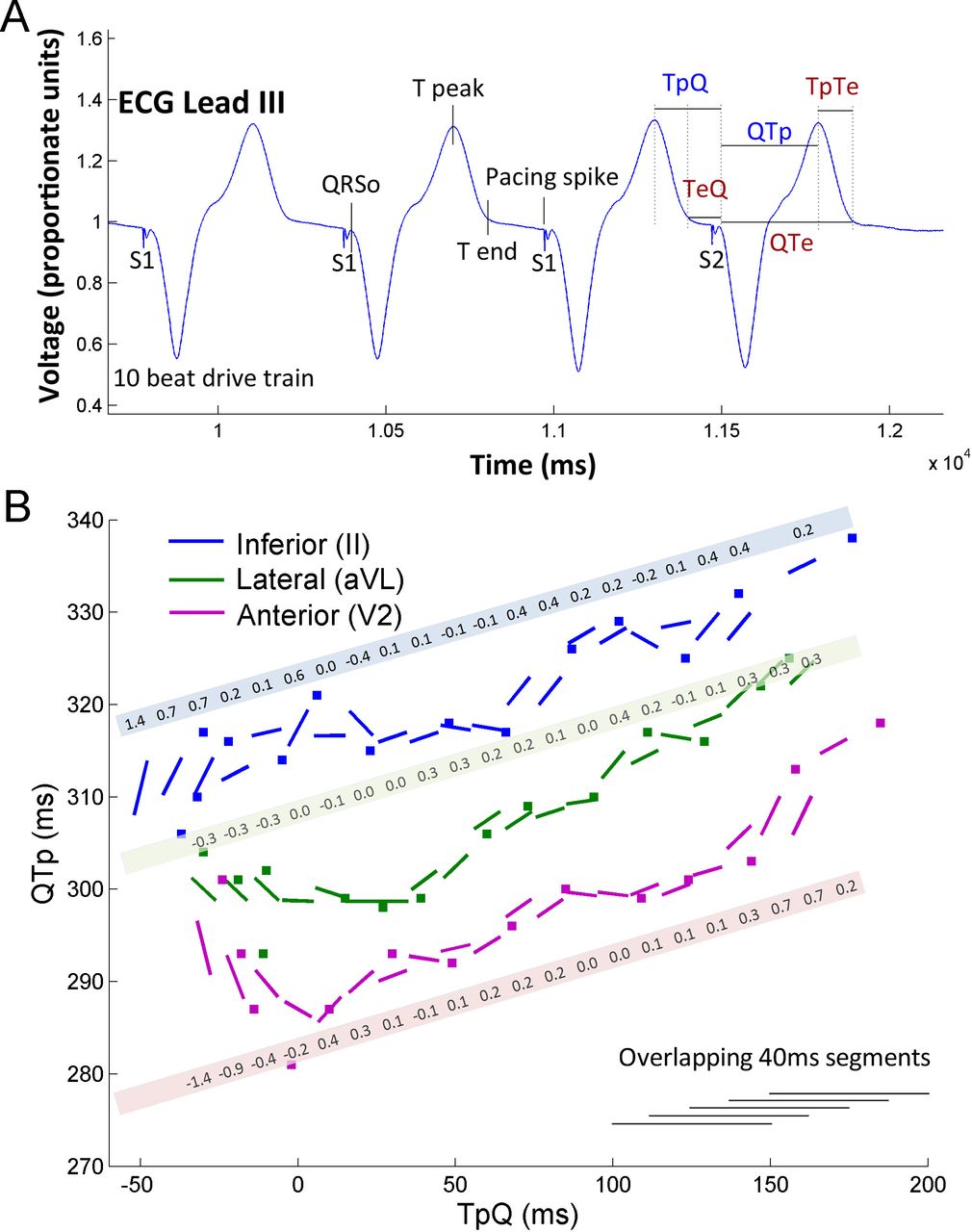
The ECGs recorded during the EP study will be exported at 16-bit resolution for analysis. The digital ECG data will be transferred to the core lab for analysis using custom software written in MATLAB® (Mathworks, Natick, USA). All data analysis and calculation of the R2I2 and PERS values will be performed by an investigator blinded to the clinical endpoints. A stepwise linear-fitting method, which is a standard approach, will be used to construct restitution curves using surface ECG surrogates for APD and DI (Q−Tpeak interval and Tpeak−Q interval) as described in previous works (figure 3).10 19 For each lead of the surface ECG, the Q−Tpeak is plotted against the Tpeak−Q. Gradients are fitted for each 40 ms overlapping least square linear segment. For each lead, in each 40 ms segment, the difference of the gradient from the mean gradient is calculated. The mean of the SD is taken as the R2I2. The mean of the peak restitution curve slope is calculated to be the PERS value.
{kind=link}
{kind=link}
{kind=link}
Derivation of Regional Restitution Instability Index (R2I2) and Peak ECG Restitution Slope (PERS). (A) Stimulation protocol demonstrating the fiducial points of TpeakQ and QTpeak (blue) which are required to plot on the restitution curve (B) gradients are fitted for each 40 ms overlapping least square linear segment. The mean of the SD of gradient differences from the mean gradient is taken as the R2I2. The mean of the peak restitution curve slope is calculated to be the PERS value (reproduced with permission from Nicolson 2014).19
Statistical considerations
Digital ECG data obtained from the EP study will be securely transferred to the Core Lab for prospective analysis and calculation (blinded to clinical outcome) of R2I2 and PERS. Once 12-month and 18-month endpoint assessment have been made for each participant, the study groups will be divided into those reaching endpoint and those not reaching endpoint. Based on existing data, R2I2 data are expected to be parametric and PERS data non-parametric. Parametric data will be expressed as mean (±SEM) and analysed using a Student’s t-test; non-parametric data as median (IQR) and analysed using the Mann-Whitney U test. Proportions will be analysed using a two-sided Fisher’s exact test. Previous work18 19 has found that an R2I2 value of 1.03 and PERS value of 1.21 provide the best ‘cut-off’ values to partition patients into ‘high’ and ‘low’ risk groups, from which Kaplan-Meier survival curves can be drawn for patient subgroups partitioned by this R2I2 cut-off and for patient subgroups partitioned by combinations of R2I2 and PERS cut-offs; comparison of cumulative endpoints will again be based on logarithmic transformations and survival will be recorded as time to first endpoint or the end of follow-up.
Sample size
The sample size was informed by a two-sample t-test power calculation using the Satterthwaite approximation for unequal variances and using the R2I2 data from a previous study (R2I2 in VA/SCD group compared with non-VA/SCD, mean±SD 1.11±0.36 vs 0.84±0.27).19 To achieve over 90% power at a 5% significance level requires a minimum of 22 patients reaching endpoint. The endpoint rate is estimated to be 5% based on Multicenter Automatic Defibrillator Implantation Trial: Reduce Inappropriate Therapy (MADIT-RIT) study data.23 Hence, to achieve 22 endpoints during 12 months of follow-up, a sample size of 440 participants will be required. A p value of <0.05 will be considered to be significant.
Ethics, dissemination and monitoring
The Steering Committee consists of the study PI’s (online supplemental appendix 1) who are responsible for the clinical and scientific conduct of the study and the publication of the results. In addition, the Steering Committee will review AE and serious AE. The research coordinator will prepare the endpoints for adjudication by the endpoints committee who will not have access to blinded data (online supplemental appendix 2). The results will inform the design of a definitive RCT. Dissemination will include peer-reviewed journal articles reporting the qualitative and quantitative results, as well as presentations at conferences and lay summaries.
Supplemental material
The study design and research protocol were approved by the Research Ethics Committees Northern Ireland (reference no. 16/NI/0069) and Health Research Authority (IRAS reference 186618, EDGE ID 51707) with informed consent being obtained from the subjects. The study is being conducted in accordance with UK laws, Good Clinical Practice, and the Declaration of Helsinki 2002.
Discussion
LVEF is the current predominantly used, lealst-worst tool for ICD risk stratification. The reliance on this marker leads to a large number of patients who, on the basis of LVEF, are considered low risk but go on to have SCD. This is while a substantial proportion of patients receiving ICDs do not make use of them; this results in considerable, unnecessary cost and morbidity. In the Multicenter Automatic Defibrillator Implantation Trial II (MADIT-II) and Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) trials, in which reduced left ventricular (LV) function was the main marker of risk, only 10% of patients received appropriate ICD shock therapy per year during the 4-year follow-up period.4 5
Basic science research on electrical restitution has been extended into translational work that has led to the development of two novel risk markers of SCD. R2I2 and PERS represent a technology using familiar ECG recording equipment and can be performed with minimal specialist training. R2I2 and PERS are both independent of LVEF in their association with VA/SCD occurrence. This raises the possibility that R2I2/PERS will retain sufficient positive predictive value in a lower risk population and it is anticipated that it will enable reclassification of patients who are currently stratified as low or medium risk to be identified to receive ICDs to prevent SCD.
Ethics statements
Patient consent for publication
Acknowledgments
The development of R2I2 and PERS includes studies which are part of the research portfolio supported by the National Institute for Health Research Leicester Biomedical Research Centre.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors This study was conceived by GAN and WBN. All the authors were responsible for the design of the study and the acquisition of the data. All the authors were involved in the drafting of this work, final approval of the manuscript and are accountable for the work.
Funding This work was supported by a restricted grant from Heart Research UK (RG2649/15/18). GAN is supported by a British Heart Foundation Programme Grant (RG/17/3/32774). GAN and WBN are supported by a Medical Research Council Biomedical Catalyst, Developmental Pathway Funding Scheme Award (MR/S037306/1).
Competing interests The University of Leicester has applied for a patent for the R2I2 and PERS ECG markers on behalf of WBN and GAN. GAN reports research fellow support from Boston Scientific Ltd and Abbott Ltd.AM’s salary is supported by Abbott (formerly St Jude Medical).
Provenance and peer review Not commissioned; peer reviewed for ethical and funding approval prior to submission.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.