Article Text

Abstract
Introduction Elevated N-terminal pro-brain natriuretic peptide (NT-pro-BNP) after non-cardiac surgery is a strong predictor for cardiovascular complications and reflects increased myocardial strain. NT-pro-BNP concentrations significantly rise after non-cardiac surgery within the first 3 days. Levosimendan is a potent inotropic drug that increases calcium sensitivity to cardiac myocytes, which results in improved cardiac contractility that last for approximately 7 days. Thus, we will test the effect of a pre-emptive perioperative administration of levosimendan on postoperative NT-pro-BNP concentration as compared with the administration of a placebo in patients undergoing moderate-risk to high-risk major abdominal surgery.
Methods and analysis We will conduct a double-blinded prospective randomised trial at the Medical University of Vienna, Vienna, Austria (and potentially a second centre in Germany), including 230 patients at-risk for cardiovascular complications undergoing moderate- to high-risk major abdominal surgery. Patients will be randomly assigned to receive a single dose of 12.5 mg levosimendan versus placebo after induction of anaesthesia. The primary outcome will be the postoperative maximum NT-pro-BNP concentration between both group within the first three postoperative days. Our secondary outcomes will be the incidence of myocardial ischaemia, myocardial injury after non-cardiac surgery and a composite of myocardial infarction and death within 30 days and 1 year after surgery between both groups. Our further secondary outcome will be stratification of NT-pro-BNP values according to previously thresholds to predict mortality of myocardial infarction after surgery.
Ethics and dissemination The study was approved by the Ethics Committee of the Medical University of Vienna on 14 July 2020 (EK 2187/2019). Written informed consent will be obtained from all patients a day before surgery. Results of this study will be submitted for publication in a peer-reviewed journal.
Trial registration number NCT04329624.
- adult anaesthesia
- heart failure
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is a double-blinded prospective randomised control trial evaluating the effect of perioperative levosimendan vs placebo on postoperative N-terminal pro-brain natriuretic peptide concentrations.
Elderly patients with increased cardiovascular risk factors will be included in this trial.
Patients, healthcare providers and study personnel as well as outcome assessors will be blinded to randomisation.
This trial is not powered to evaluate the effect on myocardial injury after non-cardiac surgery, myocardial infarction, and death within 30 days after surgery.
Introduction
N-terminal pro-brain natriuretic peptide (NT-pro-BNP) is a validated biomarker to assess perioperative myocardial wall stress.1–3 NT-pro-BNP has also a high-predictive value for major cardiovascular complications after non-cardiac surgery.4 5 In detail, a meta-analysis of 2179 patients has shown that postoperative BNP values over 92 ng/L or NT-pro-BNP over 300 ng/L were the strongest independent predictors of 30-day mortality and non-fatal myocardial infarction (MI) in patients undergoing non-cardiac surgery.1 We have previously shown that NT-pro-BNP concentrations increases significantly within the first three postoperative days in relatively healthy patients as well as in patients with cardiovascular risk factors undergoing moderate- to high-risk abdominal surgery, which we assumed reflect increased myocardial strain in the postoperative period.6 7
Thus, reducing myocardial strain via strengthening of myocardial contractility might lead to a lower increase in postoperative NT-pro-BNP concentrations and might finally result in improved postoperative outcome. Levosimendan is a positive inotropic drug which binds at troponin C and increases the sensitivity for calcium during high intracellular calcium concentrations during systole. This leads to more facilitated actin-myosin complex formations which increases contractility.8 Furthermore, levosimendan improves myocardial function without increasing myocardial oxygen consumption.9 10 Levosimendan is indicated for short-term treatment in patients with acute decompensated heart failure, in which conventional treatment is not sufficient. The duration of action of levosimendan exceeds nearly 7 days after the administration of a single dose.11 In this context, we assume that, based on its favourable pharmacodynamic and pharmacokinetic profile, levosimendan might be very considerable to improve myocardial contractility in the operative setting.11
Several studies investigated the effect of levosimendan in patients undergoing cardiac surgery.12–14 The Levosimendan in Patients with Left Ventricular Dysfunction Undergoing Cardiac Surgery (LEVO-CTS), which is the most recent and largest trial, showed no significant difference in 30-day mortality, renal replacement therapy, perioperative MI or the need of a mechanical cardiac assist devise, between patients receiving levosimendan as compared with patients receiving placebo during cardiac surgery.12 However, in recently published secondary analysis of the LEVO-CTS a significant reduction in 90-day mortality has been observed in patients with isolated coronary artery bypass graft (CABG) surgery.15 Interestingly, no effect on 90-day mortality has been observed in patients having CABG combined with valve replacement surgery.15 In this context, the effect of levosimendan during cardiac surgery, is also still not entirely cleared. Even more, evidence of perioperative levosimendan administration in patients undergoing non-cardiac surgery is lacking.
Therefore, we will test our primary hypothesis that the perioperative administration of a single-dose of levosimendan will significantly attenuate the release of postoperative maximum NT-pro-BNP in patients at risk for cardiovascular complications undergoing moderate-risk to high-risk major abdominal surgery within the first three postoperative days as compared with placebo. We will also test the secondary hypothesis that levosimendan decreases the incidence of myocardial injury after non-cardiac surgery (MINS), diagnosed by consecutive troponin T measurements (assessed within 2 hours after surgery, and on the first, second and third postoperative day). Further, we will test the secondary hypothesis that levosimendan lead to a lower incidence of a composite of MI, and death within 30 days and 1 year after non-cardiac surgery. A further secondary outcome will be stratification of postoperative maximum NT-pro-BNP concentration according to previously published thresholds.1
Methods and analysis
Objectives and design
We will conduct the “Effect of periperative Levosimendan administration on postoperative N-terminal pro brain natriuretic peptide concentration in patients with increased cardiovascular risk factors undergoing non-cardiac surgery” - IMPROVE trial, a group sequential double-blinded parallel group randomised clinical trial, at the Medical University of Vienna. We will include 230 patients at-risk for postoperative cardiovascular complications undergoing major abdominal surgery. Patients will be randomised either receiving a single dose of levosimendan or placebo before induction of anaesthesia. We have followed the Standard Protocol Items: Recommendations for Interventional Trials recommendations in preparing this protocol. The study was approved by the Ethics Committee of the Medical University of Vienna on 14 July 2020. We started patient enrolment on 1 October 2020. The IMPROVE trial was primarily planned as a single-centre study. We are currently planning to involve a second study centre: Charité-Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt Universität zu Berlin, Department of Anesthesiology and Intensive Care Medicine, Campus Benjamin Franklin Hindenburgdamm 30, 12203 Berlin, Germany.
The IMPROVE trial will test the primary hypothesis that the perioperative administration of levosimendan will significantly decrease postoperative maximum NT-pro-BNP concentration as compared with the administration of placebo in patients, who are at least 45 years or older, at-risk for cardiovascular complications undergoing major abdominal surgery.
We will further test the secondary hypothesis that the administration of levosimendan will result in a lower incidence of MINS as compared with placebo. We also test the secondary hypothesis that patient receiving levosimendan will have a significantly lower incidence of the composite of MI, and death within 30 day and 1 year after surgery, as compared with placebo. We will also test the secondary hypothesis that the administration of levosimendan will result in a significantly lower WHO Disability Score (WHODAS) score assessed 30 days and 1 year after surgery as compared with placebo.
Our further secondary hypothesis is that patients receiving Levosimendan will have a lower risk of mortality within 30 days after surgery determined by stratification according to previously published thresholds.1
Outcomes
Our primary outcome will be the maximum NT-pro-BNP concentration within the first three postoperative days between both groups.
Our secondary outcomes will be the incidence of myocardial ischaemia, MINS, and a composite of MI and death within 30 days and 1 year after surgery between both groups. Our further secondary outcome will be WHODAS scores at 30 days and 1 year after surgery between both groups. Our further secondary outcome will be stratification of NT-pro-BNP values according to previously thresholds to predict mortality within 30 days after surgery.
Overview of our outcome assessments is shown in table 1.
Outcomes assessment
We will use the fourth Universal Definition to diagnose MI. MINS will be defined using the following high-sensitive troponin T thresholds: (1) troponin T of 20 to <65 ng/L with and absolute change of at least 5 ng/L or (2) troponin T level >65 ng/L. Furthermore, in patients whose troponin T concentration was adjudicated from non-ischaemic aetiology (eg, sepsis, pulmonary embolism), will be not considered as having MINS.
Study population
The inclusion and exclusion criteria are presented in box 1.
Inclusion and exclusion criteria
Eligibility
All patients need to meet all of the following criteria for inclusion (1–4):
Major surgery planned for more than 2 hours.
≥65 years of age and ≤85 years of age.
Provide written informed consent and.
Fulfil ≥2 of the following criteria (A–E).
Inclusion criteria (A–F)
N-terminal pro-brain natriuretic peptide ≥200 ng/L
History of coronary artery disease defined as 1 of the following 7 criteria (I to VII):
History of angina.
History of myocardial infarction or acute coronary syndrome.
History of a segmental cardiac wall motion abnormality on echocardiography/radionuclide imaging.
History of positive myocardial stress test (echocardiographic or radionuclide).
History of a coronary artery stenosis >50%.
ECG with pathological Q waves in any two contiguous leads.
History of previous artery revascularisations.
History of permanent/paroxysmal atrial fibrillation diagnosed by physician/specialist.
History of peripheral arterial disease as defined by a physician/specialist diagnosis of a current, or prior history of any 1 of the following 5 criteria (I–V)
Intermittent claudication.
Stenosis ≥70% detected by angiography or Doppler.
Stenosis ≤70% detected by angiography or Doppler and requiring medical treatment for example, acetylsalicylic acid (ASA) or other platelet inhibitor.
History of stroke or transient ischaemic attack (TIA)—diagnosed by physician or CT/MRI.
Diagnosed cerebral arteriovascular disease diagnosed by a physician/specialist.
Any 3 of 10 of the following risk criteria (1–10)
History of congestive heart failure defined as a physician diagnosis of a current or prior episode of congestive heart failure OR prior radiographic evidence of vascular redistribution, interstitial pulmonary oedema, or frank alveolar pulmonary oedema.
History of a transient ischaemic attack.
Diabetes and currently taking an oral hypoglycaemic agent or insulin.
History of hypertension.
Hyperlipidaemia and currently taking a lipid-lowering agent.
Documented chronic kidney disease diagnosed by physician/specialist and creatinine clearance >30 mL/min.
History of smoking within 2 years of surgery.
Diastolic dysfunction (≥grade 1) documented by echocardiography.
Age ≥70 years.
Preoperative troponin T (5th generation) ≥ 25 ng/dL.
Exclusion criteria (A–J)
Previous adverse response and/or allergy to levosimendan.
Intensive care unit (ICU) patients undergoing surgery.
Preoperative sepsis/systemic inflammatory response syndrome (SIRS) needing ICU treatment.
Preoperative haemodynamically instable patients, who require vasopressor or inotropic support.
Renal or liver transplantation.
History of severe heart failure (eg, left ventricular ejection fraction (LVEF) <30%).
Patients undergoing surgery for pheochromocytoma.
Liver cirrhosis.
Diagnosed pulmonary hypertension (mean pulmonary arterial pressure (mPAP) >25 mm Hg—right heart catherisation) requiring treatment.
Severe renal failure defines as creatinine clearance <30 mL/min.
Study personal will screen a day before all planned operations. Study personnel will approach all eligible patients to obtain written informed consent a day before surgery. Previous experience showed that approximately 3–4 patients per week will meet the inclusion criteria. Therefore, we assume a period of 12–24 months to complete this trial.
Anaesthesia protocol
We will perform an ECG, blood pressure and peripheral oxygen saturation in all patients. Arterial blood pressure will be measured after induction of anaesthesia in all patients. A central venous line will be placed and monitored the discretion of the attending anaesthetist.
We will induce anaesthesia with 1–3 µg/kg body weight (BW) fentanyl, 2 mg/kg/BW Propofol and 0.6 mg/kg/BW rocuronium. We will maintain anaesthesia with sevoflurane/desflurane in a mixed oxygen carrier gas and Propofol infusion of 6–8 mg/kg BW, respectively. Sevoflurane/desflurane or propofol will be titrated adjusted using processed electroencephalography (NarcoTrend, Narcotrend-Gruppe, Hannover, Germany). Fentanyl administration will be repeated, as appropriate. When remifentanil will be used, the infusion rate will be increased until adequate analgesia will be reached. Muscle relaxation will be maintained to 1–2 mechanical twitches in response to supramaximal stimulation (train-of-four stimulation, target of <75%).
After endotracheal intubation, all patients will be mechanical ventilated using a FiO2 between 0.3 and 0.5 to maintain a SpO2 >95% or pO2 of >80 mm Hg. The tidal volume will be set between 6 and 8 mL lean BW to maintain an end-tidal CO2 concentration within 35 to 40 mm Hg. Positive end-expiratory pressure will be set at 5 mm Hg or higher according to the patients’ requirements.
We will actively warm patients with convective warming to maintain perioperative normothermia.
Fluid management during surgery will be goal-directed guided using an oesophageal Doppler (CardiacQ, Deltex Medical, Chichester, Westsussex) according to a previous published algorithms.16 17 We will use an acetate buffered crystalloid solution according to the standard of clinical care. In the case of acute bleeding volume administration according to the algorithm may not be sufficient. To keep place with fluid requirements, further volume boluses (5% albumin, 20% albumin) will be administered as necessary according to the attending anaesthetist. We will maintain a haematocrit of at least 26%.
Randomisation
Randomisation will be conducted after non-invasive blood pressure measurement is performed (within 1 hour before induction of anaesthesia). Preoperative systolic blood pressure has to be at least 110 mm Hg that the patient is able to be randomised. If the preoperative systolic blood pressure is lower than 110 mmH, the patient will not be randomised. We will use the online randomisation programme ‘Randomizer’ (Randomizer, Medical University of Graz, Graz, Austria https://www.meduniwien.ac.at/randomizer/web/login.php) provided by the Medical University of Vienna. Randomisation will be performed using permutated blocks. Random permutated blocks of size 4 or 6 are used, that is, the blocks are of different sizes and the size of the next block is randomly chosen from the available block sizes 4 or 6. There will be no stratification of the randomisation. If, however, the second study site is opened, we will stratify the randomisation by site and will also use a stratified Wilcoxon test.
Randomisation can only be performed by registered persons. For log-in, an individual user-ID and password entry are necessary. To ensure the double-blind character of the study, only the unblinded study personal, who is involved in the preparation of the study drug, but not in the data acquisition, will be able to randomise patients. Randomisation will be conducted only if patients meet all inclusion criteria (checking of inclusion and exclusion criteria; obtaining written informed consent of the patient; preoperative blood pressure measurement performed by blinded study team members). Before randomisation, the person who conducts the randomisation receives information about the ID-number of the patient that should be randomised. The ID-Number will be a consecutive number of the patients who meet the inclusion criteria. The advantage of the online randomisation with the ‘Randomizer’ is that for each patient, information will be available, when the patient was randomised and by whom (figure 1).

Study flow chart. NT-pro-BNP, N-terminal pro-brain natriuretic peptide; WHODAS, WHO Disability Score.
Intervention
Patient will be randomly assigned to receive a solution of 500 mL 5% glucose solution containing 12.5 mg of levosimendan or a solution of 500 mL containing a placebo. The study drug will be started immediately after skin incision with a rate of 0.48 mL/kg/min. In the case, that the patient will need continuous vasopressor support at the time during skin incision, we will halve the study drug infusion rate to 0.24 mL/kg/min (figure 2).

Study procedure. ICU, intensive care unit; PACU, postanaesthesia care unit.
We will adapt the study drug infusion rate according to the investigator brochure as follows:
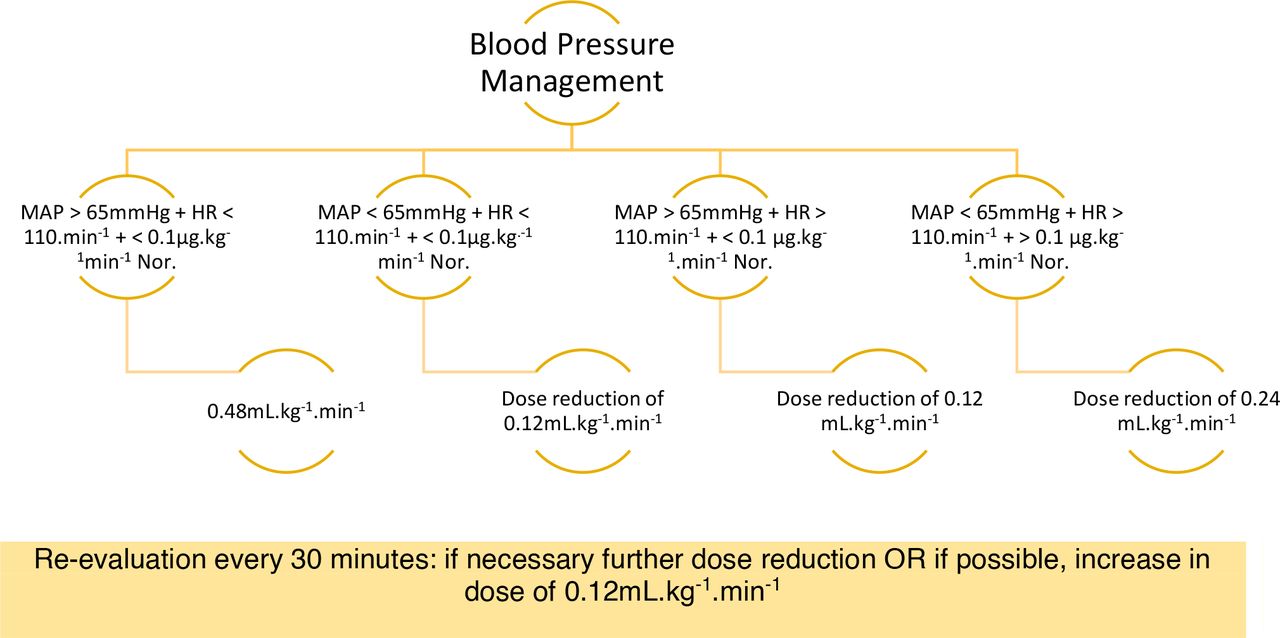
Study drug infusion will be stepwise reduced of about 0.12 mL/kg/min in the case of clinical significant hypotension (clinical significant hypotension is defined as mean arterial pressure <55 mm Hg for more than 10 min)18 or if tachycardia (defined as heart rate >110 beats per minute) occurs. We also define clinical significant hypotension when the administration of more than 0.24 mL/kg/min norepinephrine is needed to maintain an adequate mean arterial pressure (defined as mean arterial pressure >65 mm Hg) without the signs of hypovolaemia, bleeding or sepsis.
We will increase the infusion rate up to 0.24 mL/kg/min during haemodynamic stability. This is defined as mean arterial pressure within the 20% range of baseline measurements without the need of vasopressor support (figure 3).

{kind=link}
{kind=link}
{kind=link}
Recommendation for infusion rates according to haemodynamic changes.
Postoperative
We will monitor ECG, SpO2 and arterial blood pressure in all patient’s closely during the time the study drug will be administered. All patients will stay overnight at the postanaesthesia care unit (PACU) or intensive care unit (ICU) as appropriate, for further haemodynamic monitoring.
Measurements
Demographic data
We will record age, sex, body mass index, American Society of Anesthesiologists physical status (ASA), history of tobacco use, medication and medical history. We will record preoperative laboratory parameters.
Intraoperative and postoperative data
Routine intraoperative and postoperative anaesthesia specific data will be extracted from our electronic anaesthesia record system. This will include arterial blood gas analysis, time of anaesthesia and surgery, intraoperative fluid management, respiratory settings, blood loss, blood given, urinary output, intraoperative medication and haemodynamic data (including mean arterial pressure, heart rate, stroke volume, cardiac output, central venous pressure) and the use of vasopressors will be recorded. Data from the PACU and/or ICU will include blood pressure, heart rate, fluid management, urinary output and the amount of vasopressor administered.
Study-specific measurements
We will assess NT-pro-BNP, troponin T, copeptin, procalcitonin, interleukin-6 before induction of anaesthesia, within 2 hours after surgery, on the first, second, third and fifth postoperative day. Routinely performed laboratory measurements are at the discretion of the attending physician and will be recorded for the first three postoperative days as well. We measured all study specific biomarkers on the fifth postoperative day to be able to reflect the pharmacokinetic profile of the active metabolite. All measurements will be assessed by the Department of Laboratory Medicine of the Medical University of Vienna.
We will further assess the WHODAS 2.0 questionnaire before surgery, and 30 days and 1 year after surgery.
Overview of all study specific interventions are shown in table 2.
Schedule of events
Data analysis
Baseline characteristics such as age, sex, ASA physical status, revised cardiac index, tobacco and alcohol use, comorbidities, long-term medication and type of surgery, as well as laboratory values such as creatinine, haemoglobin, sodium and C reactive protein will be analysed descriptively. For continuous variables mean, SD, minimum, first quartile, median, third quartile and maximum will be calculated for both treatment groups. For categorial variables, absolute and relative frequencies will be calculated for both treatment groups.
If hospital discharge occurs before the third postoperative day, the maximum value of the available data will be used for outcome measure.
The primary hypothesis will be tested using a Wilcoxon-Mann-Whitney U test. If a patient dies before NT-pro-BNP concentration can be assessed within the third postoperative day, the patient’s NT-pro-BNP value will be set to the maximum. Maximum NT-pro-BNP concentration is defined as the highest value measured 2 hours after surgery, the first, second and third postoperative day. An interim analysis will be performed after the collection of data of a third of the overall planned patients. To control for the overall type 1 error rate of 0.05, O'Brien-Flemming boundaries for group sequential designs will be used, hence for the interim analysis the two-sided significance level is 0.0007 and for the final analysis the two-sided significance level is 0.0497.19
The secondary hypothesis, whether Levosimendan decreases maximum troponin T—which is defined as the maximum of the troponin T values measured within 2 hours after surgery and on the first, second and third postoperative day—as compared with placebo will be tested using a Wilcoxon-Mann-Whitney U test.
The secondary hypothesis, whether Levosimendan decreases the incidence of MINS during the first three postoperative days as compared with placebo, a logistic regression will be calculated, where incidence of MINS during the first 3 days is the dependent variable, and the treatment group is the explanatory variable. Furthermore, age and sex will be used as covariates.
The secondary hypothesis, whether levosimendan reduces the event rates of MI and death during 30 days after surgery as compared with placebo, will be tested with a Cox proportional-hazards model. The composite endpoint MI and death within 30 days is the dependent variable and the treatment group is the explanatory variable. Age and sex will be used as covariates.
The number of patients between both study groups grouped according to the previous evaluated NT-pro-BNP thresholds will be analysed with a Wilcoxon rank-sum test.
For analysis of the 30-day and 1-year WHODAS, we will use a Wilcoxon rank-sum test to compare the individual scores between both study groups on each measurement time point and furthermore we will compare the total sum of all scores between both study groups using a Wilcoxon rank-sum test.
Sample size considerations
For the control group, data from a previous study were available, where NT-pro-BNP was measured within 1 hour after surgery and on postoperative day 1 and 3.7 The maximum of these values was calculated. For the calculation of the sample size, we assume median maximum NT-pro-BNP is reduced by 37%20 in the treatment group. To estimate the power, the distribution of the z-approximations of the Wilcoxon test statistics was estimated using the bootstrap method. A total of 10 000 bootstrap samples were drawn. The data for the control group were sampled from the data from the previous study.7 The data for the treatment group was sampled from the same data set, where all values were reduced by 37%. We considered a drop-out rate of 5%. With a total sample size of 230 (115 per group, with a planned interim analysis after 38 patients per group) the overall estimated power (ie, the power to reject either at interim or at the final analysis) is 80%. To correct for multiple testing due to the interim analysis, O'Brien-Flemming’s design will be used (see statistical analysis).
Assignment on intervention
We will measure blood pressure in all patients shortly before randomisation. When systolic blood pressure will be lower than 110 mm Hg, we will not randomise the patient.
Monitoring
Access to data
Any data required to support the protocol can be supplied on request.
Data safety monitoring board
The following experienced researches will compromise the Data and Safety Monitoring Board (DSMB): Andrea Kurz, MD (Department of Outcomes Research and General Anesthesiology, Anesthesiology Institute, Cleveland Clinic); Gerd Silberhumer, MD (Department for Surgery, Medical University of Vienna, Medical University of Vienna); Eva Base, MD (Department of Anaesthesia, Intensive Care Medicine and Pain Medicine, Medical University of Vienna); Michael Wolzt, MD (Department of Clinical Pharmacology, Medical University of Vienna)
The DSMB will evaluate adverse events (AEs, serious AEs (SAEs), suspected unexpected serious adverse reactions (SUSARs), adverse drug reaction (ADR)) from 76 patients (38 per group) after all data are available. It will be the responsibility of this committee to alert the local ethics committee via letter to any harmful effects in one of the study groups. This committee, along with the local ethics committee will have the exclusive authority to stop the study either for futility, harm or clear benefit. Any morbidity potentially related to the study protocol will be reported to the ethics committee.
Emergency unblinding and termination of the study drug
Emergency situations may require unblinding of treatment. As the treating physician (investigator) is responsible for the medical care provided to the trial participant, the decision to break to unblind the participant in emergency situations will lie solely with the investigator. The investigator may contact the DSMB to seek advice about unblinding. This should allow to reduce unblinding. If the primary investigator believes emergency unblinding is essential for the patient’s management then it can be undertaken. Only, if the primary investigator is not available a coinvestigator will be contacted for emergency unblinding.
In the case of unblinding, the study drug will be discontinued. Blood samples will be continued according to the study protocol for safety reasons. Patients will not be dropped out because of intention to treat.
Termination of the study
In the case of a preterminal termination of the study because of significant incidence of AEs evaluated by the DSMB the sponsor will notify the competent authorities the end of the study including an appropriate justification. If the study will be terminated because of safety reasons the European Medicines Agency will be notified as well.
Adjudication of the trial outcomes
Outcome adjudicators (a committee of clinicians with expertise in perioperative outcomes) who are blinded to treatment allocation will adjudicate the following outcomes: heart failure, MI, life-threating, new onset of atrial fibrillation, sepsis, stroke/TIA, pulmonary embolism, unplanned ICU admission and death. We will use the decisions of the outcome adjudicator for all statistical analysis of these events. Barbara Kabon, MD (Medical University of Vienna) and Giovanna Lurati, MD (University Düsseldorf, Germany) will chair the Adjudication Committee. Definitions for outcome adjudication are shown in the online supplemental appendix 1. Outcomes will be adjudicated in regular intervals (at least after every 30 patients). All outcomes will be reviewed by both outcome adjudicators.
Supplemental material
Reporting of SAEs
Adverse (drug) reaction
All noxious and unintended responses to a medical product related to any dose should be considered as an ADR. ‘Responses’ to a medical product means that a causal relationship between a medical product and AE is at least a reasonable possibility, that is, a relationship cannot be ruled out.
The following guidelines will be used to describe severity of the AE:
Mild
Events require minimal or no treatment and do not interfere with the participant’s daily activities.
Moderate
Events result in a low level of inconvenience or concern with the therapeutic measures. Moderate events may cause some interference with functioning.
Severe
Events interrupt a participant’s usual daily activity and may require systemic drug therapy or other treatment. Severe events are usually potentially life threatening or incapacitating. Of note, the term ‘severe’ does not necessarily equate to ‘serious’.
Suspected unexpected serious adverse reactions
SUSARs are events that meet the following criteria:
Suspected to be causally associated with the study medication.
Serious (as defined for an SAE).
The nature or severity of the event is not consistent with the applicable reference safety information.
Serious AEs
An AE or adverse reaction is considered serious if, it results in any of the following outcomes:
Death.
A life-threatening experience.
A persistent or significant disability/incapacity
A congenital anomaly/birth defect
Requires hospitalisation or prolongation of existing hospitalisations.
NOTE: Any hospital readmission with at least one overnight stay will be considered as an inpatient hospitalisation. An emergency room visit without hospital admission will not be recorded as an SAE under this criterion, nor will hospitalisation for a procedure scheduled or planned before signing of informed consent. However, unexpected complications and/or prolongation of hospitalisation that occur during elective surgery should be recorded as AEs and assessed for seriousness. Admission to the hospital for social or situational reasons (ie, no place to stay, live too far away to come for hospital visits) will not be considered inpatient hospitalisations.
An important medical event.
Efficacy or safety outcomes will not be considered as SAEs, except if, because of the course or severity or any other feature of such events, the investigator, according to his/her best medical judgement, will consider these events as exceptional in this medical condition. All events considered as part of the primary, secondary or exploratory outcomes, should be reported on the appropriate page(s) in the case report form (CRF’s) but not as an SAE, unless considered exceptional in this medical condition. All AEs will be recorded on the appropriate CRF.
Only unexpected and not previously described SAEs that are believed with a reasonable level of certainty to be associated with the trial medication need to be reported immediately (ie, within 24 hours of knowledge of the event) to the sponsor of the study, the Medical University of Vienna. For such events research personal should complete an SAE CRF immediately. Research personnel in representation of the sponsor will then inform regulatory authorities in a timely manner. The DSMB will provide oversight of patients’ safety throughout the trial by reviewing unblinded aggregated data.
Data management and safety
All hard-copy forms, such as CRFs, source data and informed consents will be stored in locked rooms within a secured area and is only accessible by investigators involved in the trial.
For electronic data management we will use the CLINCASE software (Quadratek, Berlin, Germany). Data storage, back up and hosting of the CLINCASE software will be provided by IT Systems & Communications—IT4Science (Medical University of Vienna). Access to data is strictly controlled and will only be provided to the Sponsor (Medical University of Vienna), the study investigators, ethic commission (Ethic commission of the Medical University of Vienna) and if requested the Austrian Competent Authorities (Bundesamt für Sicherheit im Gesundheitswesen (BASG). Orion pharma will have no access to the data at any time.
All entries and changes in the CLINCASE software will be tracked.
Data will be stored after publishing of the trial and all of the substudies by Iron Mountain Austrian Archivierungs (Gewerbeparkstraße 3, 2282 Markgrafneusiedl, Austria) for a period of not less than twenty-five years in accordance to the Conduct of a Clinical Trial (ICH E6 Section 8) and as required by the national laws.
Patient and public involvement
No patient or public involvement.
Ethics and dissemination
Ethics approval was obtained from the ethics committee of Medical University of Vienna on 14 July 2020 (EK 2187/2019). The study was approved by the Austrian Federal Office for Safety in Healthcare (BASG) and is registered at ClinicalTrials.gov (NCT04329624; 1 April 2020). Ethical approval will also be required from any additional study centres. An investigator will obtain written informed consent from all participants after checking eligibility (the informed consent for is provided in online supplemental appendix 2. The study will be conducted according to the Declaration of Helsinki and Good Clinical Practice.
All changes regarding the study protocol will be communicate to the appropriate relevant parties.
Results of this study are planned to be published in a peer-reviewed journal. The manuscript for publication will be the authors’ own work and professional writers are not intended to be involved.
Discussion
The aim of this trial is to evaluate the effect of a perioperative administration of levosimendan on postoperative NT-pro-BNP concentrations in patients at-risk for cardiovascular complications undergoing moderate to high-risk major abdominal surgery within the first three postoperative days.1 Furthermore, we will evaluate the effect of levosimendan on the incidence of MINS, MI, and death within 30 days and 1 year after surgery.
In two of our previous trials, we observed a nearly fourfold increase in NT-pro-BNP concentration within the first 3 days after moderate-risk to high-risk major abdominal surgery in patients at-risk for cardiovascular complications and even more interestingly this significant increase was also observed in relatively healthy patients.7 21 It is known that postoperative maximum NT-pro-BNP concentration within the first three postoperative days is a strong predictor for postoperative cardiovascular events.1 22 In detail, it has been shown that the highest NT-pro-BNP value within the first 3 days is associated with postoperative mortality, cardiac mortality and nonfatal MI, and cardiac failure at both 30 days and 180 days or more after surgery.1 It was suggested by the authors that this significant increase in postoperative NT-pro-BNP concentration is and indicator for the development of postoperative subclinical heart failure.1
Although the perioperative administration of levosimendan has been widely tested in patients undergoing cardiac surgery and in patients with decompensated heart failure, no data are available for non-cardiac surgical population.12 20 Interestingly, although the study by Mehta et al found a lower incidence of low cardiac output syndrome and secondary need of inotropic use in patients in the levosimendan group compared with the placebo group, there was no reduction in 30 days mortality in the levosimendan group.12 The study in the nonsurgical setting, which specifically evaluated the effect of levosimendan in patients with decompensated heart failure showed a nearly 40% reduction in NT-pro-BNP concentration as compared with the administration of dobutamine.20 Interestingly, however, there was no significant difference in survival within 180 days.20 An important difference between our trial and the trial by Mebaza et al will be, that we will start the administration of levosimendan early during the course of surgery in a preemptive way before myocardial strain, specifically during at the postoperative period will occur. We will measure the extend of postoperative myocardial strain on the basis on the maximal rise in NT-pro-BNP concentration within the first 3 days.1 We have previously shown that NT-pro-BNP concentrations did not increase immediately after surgery, but within the first three postoperative days, which we assumed might be an indicator of myocardial strain.7 21
Although there are several positive inotropic drugs available, we will use levosimendan because of its unique pharmacokinetic profile. In contrast to dobutamine or milrinone, which has to be administered continuously and requires tight blood pressure monitoring, the active metabolite of levosimendan has a half-live of about 80 hours.11 Furthermore, it has been shown that the administration of levosimendan is save and is not associated with severe haemodynamic perturbations even in patients with acute decompensated heart failure.20 Specifically, only a slight decrease in systolic blood pressure of approximately 5 mm Hg after starting levosimendan has been observed.20 Therefore, we consider levosimendan to be an effective drug for perioperative myocardial optimisation which can easily be integrated into clinical routine care.
This study will have some limitations. First, our primary outcome will postoperative maximum NT-pro-BNP concentration between both groups. The study is not powered to evaluate the effect on morbidity and mortality. We will evaluate the incidence on MINS, MI, and death within 30 days after surgery as our secondary outcomes. Therefore, the results of these outcomes can only be used with some limitations. Furthermore, our patient population is limited to elderly patients with comorbidities. Therefore, the generalisability on younger patients with cardiovascular risk factor will be limited.
Trial status
Actual protocol V.2.4; 12 September 2021. Recruitment started 1 October 2020.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors CR, BK, EF and AG: conceptualisation and study design. CR, BK, EF and AG: first draft of the manuscript. CR, AT, NA, KH, MK, CB and ST: data collection. AT, NA, MF, AG, ST and CB: data management. CR, BK, EF, AT, NA, KH, MK, AG, MF, ST and CB: editing and critical review of the manuscript. All authors read and approved the final manuscript.
Funding The study was funded by the Medical University of Vienna. The study drug, including preparation and labelling of levosimendan and placebo, will be provided by Orion Pharma (Orion Corporation, Espoo, Finland).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.