Article Text

Abstract
Introduction Sepsis and septic shock have mortality rates between 20% and 50%. In sepsis, the immune response becomes dysregulated, which leads to an imbalance between proinflammatory and anti-inflammatory mediators. When standard therapeutic measures fail to improve patients’ condition, additional therapeutic alternatives are applied to reduce morbidity and mortality. One of the most recent alternatives is extracorporeal cytokine adsorption with a device called CytoSorb. This study aims to compare the efficacy of standard medical therapy and continuous extracorporeal cytokine removal with CytoSorb therapy in patients with early refractory septic shock. Furthermore, we compare the dosing of CytoSorb adsorber device changed every 12 or 24 hours.
Methods and analysis It is a prospective, randomised, controlled, open-label, international, multicentre, phase III study. Patients fulfilling the inclusion criteria will be randomly assigned to receive standard medical therapy (group A) or—in addition to standard treatment—CytoSorb therapy. CytoSorb treatment will be continuous and last for at least 24 hours, CytoSorb adsorber device will be changed every 12 (group B) or 24 hours (group C). Our primary outcome is shock reversal (no further need or a reduced (≤10% of the maximum dose) vasopressor requirement for 3 hours) and time to shock reversal (number of hours elapsed from the start of the treatment to shock reversal).
Based on sample size calculation, 135 patients (1:1:1) will need to be enrolled in the study. A predefined interim analysis will be performed after reaching 50% of the planned sample size, therefore, the corrected level of significance (p value) will be 0.0294.
Ethics and dissemination Ethics approval was obtained from the Scientific and Research Ethics Committee of the Hungarian Medical Research Council (OGYÉI/65049/2020). Results will be submitted for publication in a peer-reviewed journal.
Trial registration number NCT04742764; Pre-results.
- intensive & critical care
- adult intensive & critical care
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
It is a prospective, randomised, controlled, multicentre study with a relatively homogeneous group of patients.
Instead of the internationally criticised hard endpoints in sepsis trials, physiological outcomes were chosen as our primary endpoints.
Shock reversal has not been used as a primary outcome in randomised trials before, therefore, sample size calculation was based on a heterogeneous population of patients with sepsis from a limited number of studies.
For safety measures we decided to treat patients in both CytoSorb-treated groups for at least 24 hours—according to current practice—therefore, we will not able to assess sustained shock reversal without haemadsorption therapy during the first 24 hours.
Background
Sepsis and septic shock are devastating conditions with mortality rates between 20% and 50%.1–3 Sepsis has an outstandingly complex pathophysiology, therefore, the clinical presentation of sepsis is often diverse and unpredictable.4 5 The process begins with the host’s immune response triggered by various insults.6 This response becomes uncontrolled and an imbalance occurs between proinflammatory and anti-inflammatory mediators. This condition is also referred to as the ‘cytokine storm’.7 During the cascade-like inflammatory response, cytokines are released, which are a heterogeneous group of proteins, mostly in the mass range of 40 kDa.8 The theory that cytokine storm may be responsible for the observed deleterious sequence of events in sepsis, raises the pathophysiological rationale of extensive removal of circulating cytokines.9 A disturbance in vascular tone regulation also develops in sepsis: vasoplegia is thought to be a key factor responsible for the death of patients with septic shock, due to persistent hypotension.10
When standard therapeutic measures, such as adequate early resuscitation, source control and organ support fail to improve the patients’ condition, additional therapeutic alternatives, called ‘adjuvant therapies’ are applied to reduce morbidity and mortality by providing some extra help.11 Several adjuvant therapies have been tested over the decades with non-conclusive results.12–14 One of the most recent alternatives is extracorporeal cytokine adsorption with a device called CytoSorb (CytoSorbents, New Jersey, USA) that has become available in clinical practice in 2011. It is a high-flow, low-resistance cytokine adsorbent, containing specially developed polymer beads with a large adsorption surface and a spectrum of adsorption between 5 and 60 kDa.15
Over 100 case studies describing the use of CytoSorb in many clinical scenarios and in general, the effects are promising, and the treatment is well tolerated.16–18 Concerning the treatment of sepsis, clinical trials are lacking at present, and we have mainly small case series.19–22 There is also an international CytoSorb Registry, and recent data analysis on 198 patients indicated, that observed mortality (65%) was substantially better as compared with the predicted (80%–20%) and the treatment also proved to be safe.23 Furthermore, recent case series and case–control studies reported profound benefit on the outcome in patients with septic shock and treated with CytoSorb.24 25 Recently, the Adsorption of Cytokines Early in Septic Shock (ACESS trial) was published, which is the first randomised clinical trial (RCT) on CytoSorb as a stand-alone haemoperfusion treatment (ie, without continuous renal replacement therapy (CRRT)) in patients with septic shock.26 It was a proof-of-concept pilot study on 20 medical patients randomised into a CytoSorb and a standard treatment group, with cytokine adsorption initiated within the first 24 hours after the onset of septic shock. The treatment proved to be safe and resulted in a significant reduction in norepinephrine requirement and serum procalcitonin (PCT) levels in the CytoSorb group as compared with controls. In a more recent propensity-score-weighted retrospective study on more than 100 patients with septic shock requiring CRRT, when patients were weighted by stabilised inverse probability of treatment weights the results suggested that CytoSorb therapy may be associated with decreased all-cause mortality at 28 days compared with CRRT alone.27
Despite the promising case series and preliminary results, several questions need to be clarified before recommendations can be made, including the right target population, the timing and the length of a single treatment and the overall duration of the therapy. Some preliminary data are suggesting that PCT is removed by the adsorber in a time-dependent manner28 being most efficient during the first 12 hours, after which removal is negligible.
Aim of the study
This study aims to compare the efficacy of standard medical therapy (SMT, group A) and continuous extracorporeal cytokine removal with CytoSorb therapy in patients with early refractory septic shock. Furthermore, we compare the dosing of CytoSorb adsorber device changed every 12 (group B) or 24 hours (group C).
Methods and analysis
Study design
It is a prospective, randomised, controlled, three-arm, open-label, international, multicentre, phase III study with adaptive ‘sample size re-estimation’ design.
The study protocol was constructed in accordance with the Standard Protocol Items: Recommendations for Interventional Trials 2013 statement.29
Randomisation
A computer-generated random number sequence will be conducted with randomly varied multiple block sizes stratified according to the participating centres with an equal (1:1:1) allocation ratio. The medical personnel in each study centre will have credentials to access the randomisation site. On this site, the medical staff has to check all inclusion criteria and the absence of all the exclusion criteria. Patients will be recruited consecutively. After the participant was registered, the allocation appears but the following allocations and the block sizes are concealed.
Blinding
It is not possible for the staff who are providing patient care to be unaware of the group assignments after randomisation. Sham procedures for the control group would be unethical. Statisticians are blinded to treatment assignments.
Duration
Duration per patient: The study starts after randomisation. In the CytoSorb groups, measurements, blood sampling and other recordings are performed immediately after the start of CytoSorb therapy (indicated as T0). In the SMT group, T0 is defined as the first recordings after randomisation. The study period ends (Te) 12 hours after shock reversal or on day 5 after randomisation or at the time of death within this period, whichever happens first. The patients will be followed up on day 28±7 and day 90±7 after randomisation. Duration of the entire study: the planned starting date of the study is June 2021, and the planned completion date is June 2024.
Study groups
Patients eligible for the study in terms of the inclusion and exclusion criteria (defined below) will be randomly assigned to one of the three study groups after informed consent. In case the patient is unable to give consent, informed consent will be obtained from the next of kin or his/her legal guardian, information on the study and the treatment will be provided by the attending physician. Patients in group A will be treated with SMT. Patients in group B will be treated with continuous CytoSorb therapy in addition to SMT; CytoSorb device will be changed every 12 hours. Patients in group C will also be treated with continuous CytoSorb therapy in addition to standard treatment, however, CytoSorb device will be changed every 24 hours. In each group, the treatment will be continued for a minimum of 24 hours, after that until shock reversal occurs, for a maximum of 5 days or until the patient’s death (figure 1).

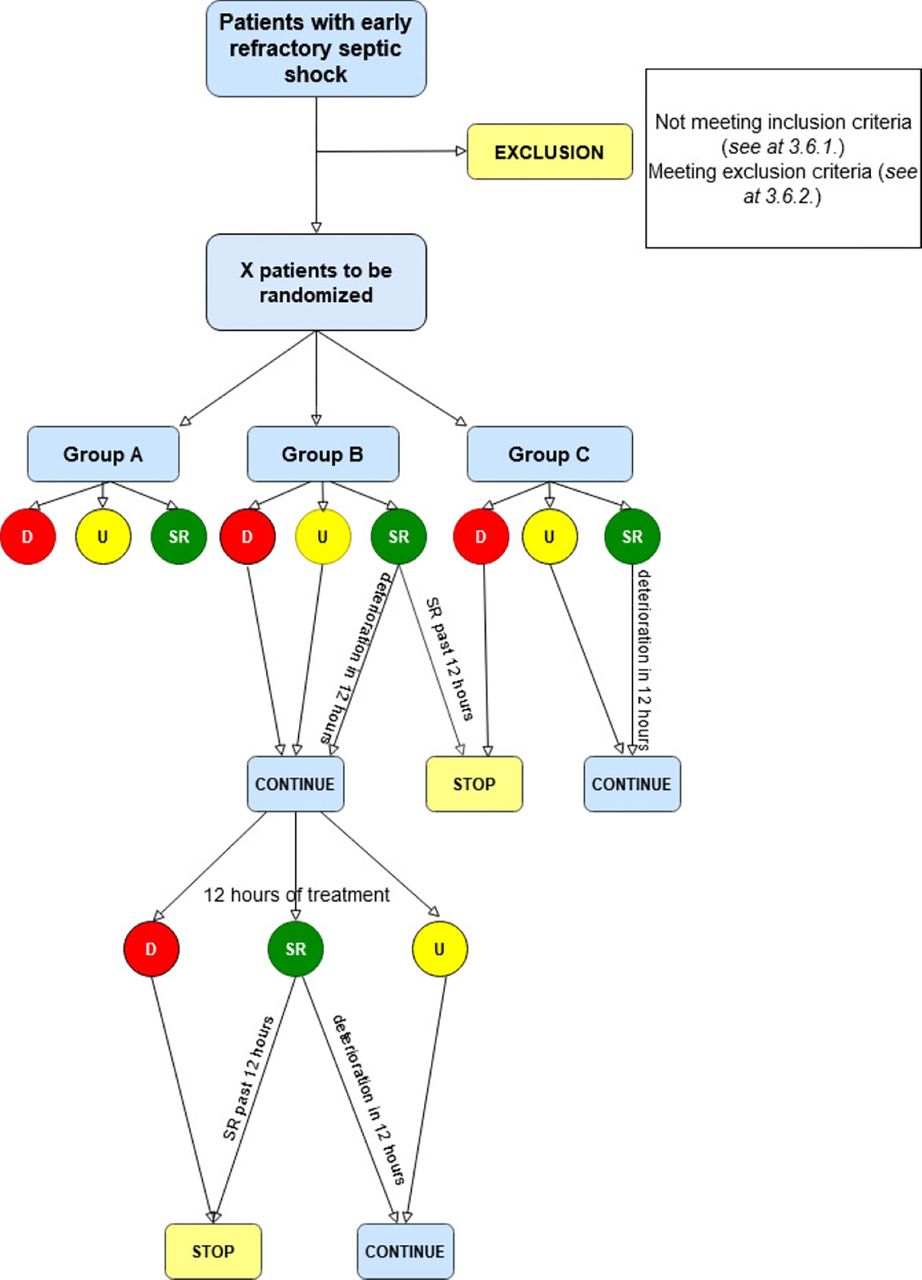{kind=link}
Flow chart of the therapy according to the SPIRIT 2013 statement.29 The figure presents 24 hours of the treatment period. D, deterioration, SPIRIT, Standard Protocol Items: Recommendations for Interventional Trials; SR: shock reversal; U: unchanged state.
Patient enrolment
The inclusion and exclusion criteria are based on the results of previous case series,24 25 on the ACESS trial26 and modified accordingly:
Inclusion criteria
Septic shock as defined by the Sepsis-3 criteria.30
Septic shock of both medical and surgical aetiology (except for reoperation).
Acute Physiology and Chronic Health Evaluation II (APACHE II) score >2524–26 (APACHE II score will be assessed at T0).
Mechanical ventilation.
Norepinephrine requirement ≥0.4 µg/kg/min for at least 30 min, when hypovolaemia is highly unlikely as indicated by invasive haemodynamic measurements24–26 assessed by the attending physician.
Invasive haemodynamic monitoring to determine cardiac output and derived variables.
PCT level ≥10 ng/mL.24–26
Inclusion within 6–24 hours after the onset of vasopressor need and after all standard therapeutic measures (including steroid therapy and/or second vasopressor) have been implemented without clinical improvement (ie, the shock is considered refractory).
Written informed consent.
Exclusion criteria
Patients under 18 years of age and over 80.
Lack of health insurance.
Pregnancy.
Criteria of standard guideline-based medical treatment not exhausted (detailed below at 3.7) SMT).
End-stage organ failure.31
New York Heart Association class IV.
Chronic renal failure with estimated glomerular filtration rate (eGFR) <15 mL/min/1.73 m2.
Model for End-Stage Liver Disease Score (>30, Child-Pugh score class C.
Unlikely survival for 24 hours according to the attending physician.
Acute onset of haemato-oncological illness.
Postcardiopulmonary resuscitation care.
Reoperation in the context of a septic insult.
Immunosuppression.
Systemic steroid therapy (>10 mg prednisolone/day).
Immunosuppressive agents (ie, methotrexate, azathioprine, ciclosporin, tacrolimus, cyclophosphamide).
HIV infection (active AIDS): HIV-VL >50 copies/mL.32
Patients with transplanted vital organs.
Thrombocytopaenia (<20.000/mL).
More than 10%-of body surface area with a third-degree burn.
Acute coronary syndrome.
In case of the need for a transfer of the patient to radiology or surgery, and if the device has to be disconnected, then the adsorber should be kept in a recirculation mode. In case of the need for changing the adsorber (ie, clotting) or if the disconnection lasted more than 2 hours, the patient should be excluded from the study.
Standard medical therapy
Patients will receive standard monitoring and care according to the centres’ local standard protocols based on international guidelines.33 It includes 5-lead ECG, pulse oximetry, continuous invasive blood pressure monitoring, central venous cannulation and advanced haemodynamic monitoring with the PiCCO-technology.
Advanced haemodynamic monitoring will be undertaken to optimise haemodynamics. Study teams will be encouraged to wean catecholamine support as soon as possible (mean arterial pressure (MAP) between 65 and 70 mm Hg in general),34 but this should remain at the physician’s discretion and should be tailored to each patient’s individual need, based on other indices of global haemodynamic parameters and tissue perfusion such as urine output, serum lactate levels, ScvO2, etc. The first choice of vasopressor is norepinephrine. For the second line, vasopressin is the recommended vasopressor—also including steroid support decided by the attending physician. In case of the need for an inotrope, dobutamine is suggested as first-line treatment. SMT will be performed according to the ’Surviving Sepsis Campaign’ Guidelines.33
Patients in both group B and C will receive a haemodialysis catheter inserted into a central vein (femoral, subclavian or internal jugular, as appropriate). Treatment will be performed as instructed by the manufacturer’s user guide.
CytoSorb therapy
In short, CytoSorb will be placed in a blood pump circuit in prehaemofilter position (haemoperfusion) using a renal replacement device—of the choice of the given site—as a stand-alone treatment or in combination with renal replacement therapy. The device will be run in continuous venovenous hemofiltration (CVVH), continuous venovenous hemodialysis (CVVHD) or continuous venovenous hemodiafiltration (CVVHDF) mode. Intravenous anticoagulation will be performed—according to the current standards recommended by the manufacturers—with heparin, low-molecular-weight heparin or citrate as required, and a pump flow rate of 100–400 mL/min will be aimed and flow rate recorded.
Physicians are strongly advised to start CytoSorb therapy as soon as possible after randomisation, but not later than 2 hours. In case of further delay, the patient should be removed from the study.
In groups B and C, special attention will be paid to coagulation, therefore, in addition to standard laboratory tests (prothrombin time, activated thromboplastin time, international normalised ratio), rotational thromboelastometry will be performed whenever necessary and available.
Antibiotic serum concentrations are recommended to be monitored—in centres where it is available—according to international standards and doses should be altered as recommended if necessary.
Shock reversal will be assessed by the attending physician and the treatment will be immediately continued or terminated with a new adsorber. Criteria for termination are as follows:
Discontinuation: shock reversal (see below) has been achieved and remains so after finishing 12 hours of SMT.26
Restarting: treatment can be restarted within 12 hours if vasopressor requirement increases despite normovolaemia confirmed with haemodynamic monitoring and in case of worsening organ function such as deterioration in gas exchange, increased extravascular lung water (EVLW), etc, which is considered by the attending physician as a result of a new onset of hyperinflammatory response.
Defining non-responders: It is expected that there will be patients who do not respond to CytoSorb treatment. Therefore, patients whose clinical condition deteriorates during and within the first 24 hours of CytoSorb therapy will be considered as non-responders and CytoSorb will not be continued. Non-responsiveness will be defined as: (A) increasing vasopressor requirement not related to hypovolaemia or bleeding, (B) increasing lactate not associated with acute liver failure and (C) when the worsening clinical picture is accompanied by increasing PCT/Interleukin-6 (IL-6) levels despite the likely presence of adequate source control.
Patients’ data will be recorded on the electronic case report form (eCRF) at T0, T6, T12, T24 and then daily until the end of the study period (Te) that is until 12 hours after shock reversal or up to a maximum of 5 days or until the patient’s death, whichever occurs first. Follow-up visits/calls are scheduled on day 28±7 and day 90±7 after randomisation.
Primary endpoints
Time to shock reversal: the hours elapsed from T0 to shock reversal.
Shock reversal: In previous studies, shock reversal occurred in 65%,24 38.5%25 and 65%26 of patients, within a 24-hour CytoSorb treatment, which has been considered as the most important clinical effect of the therapy. Based on the results ‘shock reversal’ will be defined as:
No further need or reduced (≤10% of the maximum dose) in the vasopressor requirement (including norepinephrine and/or vasopressin) for 3 hours25 35
(In case of multiple vasopressor agents are required, the reduction of one of them (≤10% of the maximum dose) is sufficient if the other agent(s)’ dosage does not need to be increased).
Low doses of vasopressor (≤10% of the maximum dose) may be required to compensate for sedation or to maintain adequate organ perfusion.
In case of (2.a) invasive haemodynamic measurements will be performed to confirm haemodynamic stability.
In case of (2.a), arterial and central venous blood gas analysis will be performed, to determine arterial lactate levels (the target is ≤2 mmol/L), venous to arterial partial pressure of carbon dioxide gap (normal value is: ≤7 mm Hg) and central venous O2 saturation (ScvO2) (increase above 70% at Te if it was lower than 70% at T0 or returning into 70%–75% by Te in case it was greater than 75%–80% at T0).
Secondary endpoints
Blood samples will be collected at T0, T6, T12, T24 and then daily, and the change from T0 to Te of the following parameters will be assessed:
Inflammatory parameters: 1. PCT, 2. IL-6, 3. C-Reactive Protein (CRP), 4. IL-1, 5. IL-1ra, 6. IL-8, 7. IL-10, 8. Tumour necrosis factor-alpha (TNF-α), 9. syndecan-1, 10. heparan sulfate.
Arterial lactate levels.
Change in Sequential Organ Failure Assessment (SOFA) score from T0 to Te (SOFA score will be assessed at T0, T24 and then daily).
Change in EVLW from T0 to Te.
Duration of mechanical ventilation in days (every 24 hours when the patient required the organ support therapy counts as one).
Duration of catecholamine requirement in days.
Duration of renal replacement therapy in days.
Need for dialysis on day 28±7.
Need for dialysis on day 90±7.
Length of stay at the Intensive Care Unit (ICU).
Length of stay at the hospital.
Survival: ICU.
Survival: hospital.
Survival at day 28
Survival at day 90.
Survival: number of days (every finished 24 hours counts one).
Adverse events (AEs).
AEs and serious AEs: definition and recording
AEss will be collected from the start of the intervention period until follow-up.
All AEs and device deficiencies including all serious AEs (SAEs) are collected and documented in the source document and the AE report form (see at online supplemental file, AEs) during the entire study period, that is, from the patient’s informed consent until the last follow-up visit/call. Dates of the event, the seriousness of the event and the relationship to the study device need to be documented. The AE report form has to be forwarded to the SC and the independent data management board (IDMB). Provided that the AE is confirmed by the SC, the national ethics committee needs to be notified (http://www.ett.hu/tukeb. htm).
Supplemental material
Follow-up
A follow-up assessment will be conducted 28±7 days and 90±7 days after randomisation using a follow-up letter/email or a phone call. In case the patient or the next-of-kin cannot be reached, medical records will be used to obtain the needed information. At day 28 and 90 survival, need for dialysis and AEs will be assessed.
Statistical analysis
Sample size calculation
Based on the previous case series and the ACESS pilot data, the most apparent clinical benefit is expected to be the reduction in norepinephrine requirement; therefore, we chose shock reversal as the most important outcome.24–26 In the ACESS trial, it was found that one single 24-hour treatment resulted in an almost 70% reduction in the required norepinephrine dose. A similar observation was made in a recent case series,24 in which a 50% reduction was found after a 24-hour treatment. Furthermore, in our pilot study, the most profound effect occurred within the first 12 hours of treatment, as far as norepinephrine requirement and PCT-level reduction are concerned.28 Based on these results, it is postulated that cytokine removal may be most effective in the first hours of treatment, therefore, shock reversal could occur faster in group B as compared with group C and faster in both groups as in group A (controls).
The sample size calculation was based on patient data from the study of Kogelmann et al.25 The time of shock reversal was separately calculated for those in whom the first adsorber was changed after 12 hours (n=3), and for those who received therapy for 24-hours each time (n=17) (48±30 hours vs 68±21, respectively). In a recent prospective RCT on patients with sepsis and septic shock, vasopressors were weaned in 96±40 hours in the control group (n=50).36
We considered these differences as clinically relevant and not to be overlooked between the three groups. Sample size calculation suggests that 135 patients (1:1:1) will need to be enrolled (45 in each study arm) to confirm or reject the hypothesis for the primary endpoint with a 20% drop-out, 80% power and 95% significance level. Non-responders will be handled as dropouts and will continue to receive SMT.
Analysis plan and statistics
Descriptive statistics—mean, median, SD, quartiles and relative frequency—weighted generalised linear model with contrasts (continuous variable) for the primary endpoint and mixed models (continuous variable), a weighted generalised linear model with contrasts (continuous variable), relative risk (dichotomous variables) for secondary endpoints. Affiliated statistical analyses will be performed with an error probability of 0.0294 (type I error probability) for per-protocol (PP) and intention-to-treat population. All statistical analyses are performed with R (V.3.5.2).
Interim analysis
Appropriate sample size calculation was not possible due to the lack of available high-quality clinical data.25 Therefore, it is highly likely that the event rate of shock reversal will occur in substantially less than 100%. In order to adapt the required sample size to maintain statistical power, we decided to allow sample size re-estimation after an interim analysis at the 50% recruitment rate. If no more subjects are needed, early termination will be applied. For this reason, the p value should be adjusted to diminish the probability of type I error; therefore, the corrected level of significance (p value) will be 0.0294.
The following rules will be applied:
If the treatment in any of the groups proves to be significantly (p<0.0294) less effective than the others and it is already obvious that there is no hope for ascertaining a significant difference between the other two groups, the study will be stopped.
If the treatment in any of the groups are significantly (p<0.0294) less effective than the others and it is already visible that there is hope of ascertaining a significant difference between the other two groups, the inferior treatment will be dropped, and the study will be continued with the remaining two arms.
If any of the groups proves to be significantly (p<0.0294) more effective than the others, the study will be discontinued.
Study populations
Safety analysis set (all patients enrolled in the study), PP Set (PPS, all enrolled patients who finished the study conforming to the requirements of the study protocol) and ITT (all randomised participants who start on a treatment, excluding consent withdrawals) will be performed.
Withdrawal of a subject from PPS
Patients will not be included in the per-protocol analysis if: (1) during the trial any exclusion criteria is met; (2) a serious adverse effect occurs; (3) data required for the primary endpoints are missing; or (4) serious medical conditions not related to septic shock occur (eg, myocardial infarction, stroke); (5) commencement of CytoSorb more than 2 hours after randomisation and (6) the duration of CytoSorb therapy did not reach 24 hours or the patient died within 24 hours from enrolment in groups B and C.
Patient and public involvement
Patients or the public were not involved in the design, conduct, reporting or dissemination plans of our research.
Ethics and dissemination
Ethical and legal considerations
This clinical study will be conducted following the Declaration of Helsinki. It will be conducted in compliance with the protocol, Good Clinical Practice (GCP) (2001/20/EEC, CPMP/ICH/135/95), designated standard operating procedures, and local laws and regulations relevant to the country of conduct. This protocol in its current version was approved by the Scientific and Research Ethics Committee of the Hungarian Medical Research Council (OGYÉI/65049/2020).
Data management
IDMB will handle data, eCRF will be applied. The investigator will guarantee that the data in the eCRF are accurate, complete and clear. Data management plan will detail the data handling during and after the trial. Data from completed eCRFs will be assessed under the direction of the data manager at IDMB according to a data cleaning plan. In case of missing, improbable or inconsistent data in the eCRFs will be referred back to the Investigator using a data query form.
Publication policy
Centres recruiting more than 10 patients can nominate two authors to the authorship list. Every additional 10 patients will give the opportunity to nominate an additional author.
Trial organisation, committees and boards
DECRISS is designed and coordinated by the Centre for Translational Medicine at the Medical School of University of Pécs.
Steering committee
The steering committee (SC) will be led by ZM (intensive care specialist). The members will be AK (medical doctor, full-time employee on the project), MM (intensive care specialist), KKo (intensive care specialist), LS (intensive care specialist), BE (clinical research specialist) and PH (clinical pharmacologist). SC will discuss all important questions including AEs and the drop-outs during the study.
Participating centres
The trial will start in two centres (University of Pécs, Pécs, Hungary; Poznan University of Medical Sciences, Poznan, Poland), then the trial is open for other centres. The centre will be assessed by the IDMB and will be presented to the SC. The SC has the right to decide whether the centre meets the required quality to join the study. Compulsory requirements for a centre are: (1) it needs to treat at least 50 patients with septic shock a year; (2) it needs to have all the equipment required for the study; (3) besides the regular medical team, the centre has to have human resources (doctors, nurse/administrator) available for the trial; and (4) before study commencement a meeting will be held; at least one person/centre needs to attend who completed a GCP course. All the details of the study protocol will be discussed thoroughly. A letter of intent needs to be sent to the corresponding author by email in case of a centre wishing to participate in the study.
Discussion
To our best knowledge, this is the first multicentre clinical trial, assessing the dosing of CytoSorb treatment alone as well as in combination with standard CRRT and compared with standard treatment in patients with refractory septic shock.
Strengths and limitations of the study
Study design intends to aim a relatively homogeneous group of patients in order to overcome the drawbacks of previous large sepsis trials, that resulted in non-significant findings.37 38 Therefore, in addition to the broad term Sepsis-3 definition of septic shock,30 other prerequisites will be incorporated into the inclusion criteria such as the minimum APACHE II score, norepinephrine dose, PCT levels, mechanical ventilation, etc.
Most sepsis randomised trials applied hard endpoints to evaluate the effects of a single treatment, such as mortality, length of hospital stay or ventilator and vasopressor-free days.39 40 However, this approach has been criticised by several internationally acknowledged experts for numerous reasons.41 42 One of the possible solutions is to design trials with physiologic primary endpoints.41 CytoSorb therapy has been shown to reduce the need of vasopressor support in several case series and studies.24 25 38 Therefore, we decided to choose ‘shock reversal’ as our primary outcome measure. Furthermore, it is not only the occurrence of shock reversal but the ‘time to shock reversal’ from the start of treatment that is of particular interest in the current study.
The current practice of applying one adsorber for 24 hours is an arbitrary one, based on the company’s recommendation and theoretical considerations. Nevertheless, several centres change the cartridge earlier (most often after 12 hours), based simply on their experience, but no study investigated this issue yet. Therefore, the current study should have important results to determine if there is any difference in the effects when the adsorber is ‘fresh’ as compared with its later performance. For this purpose, we designed a three-arm trial comparing standard therapy to 12 and 24 hours CytoSorb adsorber changing strategies to assess, which leads to faster shock reversal.
Another strength of our study is that in addition to well-acknowledged parameters indicating organ dysfunction a specific issue in the current trial will be the investigation of the evolution of EVLW during the treatment. EVLW is an indicator of increased pulmonary capillary permeability, often due to systemic inflammation.43 There is one case report indicating that CytoSorb therapy may have protective effects on vascular barrier function.44 As mechanical ventilation is also an inclusion criterium, our study may provide further insight into the relationship between cytokine removal and pulmonary function.
Although it has been shown in several experimental models that CytoSorb removes cytokines but clinical data, especially from prospective randomised trials are missing. An array of inflammatory markers and mediators are planned to be determined during the study, which can provide a further understanding of the removal properties of the device.
One of the limitations of the study is that shock reversal per se has not been used as a primary outcome, therefore, sample size calculation was based on data from a limited number of patients and a heterogeneous population of patients with sepsis. Another potential limitation is the heterogeneity of the study population. Patients with septic shock both due to medical and surgical origin will be included, while the inflammatory response might be different in the two groups.45 However, currently available clinical data indicate that both patient populations can benefit similarly from the therapy.25 Another concern regarding heterogeneity could be that CytoSorb treatment will be applied on its own as haemoperfusion and in combination with CRRT. However, we have no data yet, neither pro nor con that these two therapies interact in any way. For safety measures, we decided to treat patients in both CytoSorb-treated groups for at least 24 hours—as precurrent practice—therefore, we will not be able to assess sustained shock reversal after 12 hours during the first 24 hours.
Ethics statements
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AK, ZMo and KKo constructed the trial. LS, MLNGM, PH, KS, JS, ZMá and KKu offered recommendations and will regularly follow the study. AK, ZMo, NZ and BE outlined the manuscript, while all the authors edited the manuscript. AK prepared the figure. The sample size calculation was carried out by NG. The treatments will be carried out by ZMá, TK, KKu, KS, JS and KKo. The final manuscript was reviewed and authorised by all of the authors.
Funding This is an investigator-initiated study, without any financial support from CytoSorbents. Center costs (IT, biostatistics, trial organisation, etc) are covered by the University of Pécs, Medical School, GINOP-2.3.2-15-2016-00048 - STAY ALIVE' and GINOP-2.3.4-15-2020-00010 Competence Center for Health Data Analysis, Data Utilisation and Smart Device and Technology Development at the University of Pécs).
Competing interests ZMo is one of the medical directors at CytoSorbents Europe.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.