Article Text

Abstract
Introduction Bile leakage is a frequent complication after liver resection associated with the need of interventional drainage, endoscopic retrograde cholangio pancreatography (ERCP) or reoperation. The intraoperative application of the white test could be a promising strategy to reduce the occurrence of bile leakages. Therefore, we propose to conduct the first multicentric randomised controlled trial with rate of postoperative bile leakage as primary endpoint with and without the white test.
Methods and analysis The Bile-Leakage Trial trial is an investigator-initiated randomised controlled, parallel group, double-blinded, multicentric, superiority trial in four Swiss centres. A total of 210 patients undergoing a resection of at least 2 liver segments will be randomly allocated intraoperatively to either the intervention (identification of open bile ducts with administration of 20–40 mL SMOFlipid5% in the bile tract) or the control group (identification with a white gauze on the liver resection surface).
Primary outcome will be the comparison of the postoperative bile leakage rate in both groups within 30 days after liver resection, defined according to the classification of the International Study Group of Liver Surgery. Secondary outcomes will be operative and postoperative complication, including severity grade of the bile leakage, rate of ERCP, interventional drainage, morbidity, intensive care unit stay, and mortality.
Ethics and dissemination The cantonal ethics committees of all participating centres and Swissmedic approved the study. SMOFlipid20% consists of a mixture of oils, no side effects resulting from the intraoperative application of 20–40 mL in the biliary tract with consecutive enteral absorption are expected nor are side effects described in the literature. SMOFlipid20% will be diluted intraoperatively with isotonic saline solution to a concentration of 5%. The results of the BiLe-Trial will be submitted to a peer-reviewed journal regardless of the outcome. As this is an investigator-initiated trial, data are property of the sponsor investigator and can be obtained on request.
Trial registration number Clinicaltrials.gov, ID: NCT04523701. Registered on 25 August 2020.
Swiss National Clinical Trials Portal (SNCTP), ID: SNCTP000004200. Registered on 20 January 2021.
Protocol version V3.2_14-12-2020_clean.pdf
- surgery
- hepatology
- hepatobiliary surgery
- hepatobiliary tumours
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Postoperative bile leak is a relevant clinical problem in liver surgery.
First multicentric, superiority, double-blinded randomised controlled trial.
The intervention tests is simple, easily repeatable and standardised in all four study centres.
Primary endpoint: the incidence of 30d postoperative bile leak. Secondary endopoints: severity of the bile leakage, rate of endoscopic retrograde cholangio pancreatography, interventional drainage, morbidity, intensive care unit stay and mortality.
The resection technique and type of drainage are not standardised between surgical teams.
Introduction
Bile leakage is a frequent complication after liver resection, often with mild-to-severe clinical impact leading to the need of interventional drainage, endoscopic retrograde cholangio pancreatography (ERCP) or even reoperation. Many large series have reported an incidence ranging from 3.6% to 33%.1–5 In a recent study by Martin et al 2018 including 6859 patients, the rate of postoperative bile leakage was found to be 7.7% after any liver resection, with a statistically significant higher rate after major liver resection compared with minor resections, 12.6% versus 5.1% (p<0001), respectively.6
Strategies leading to a reduction of the rate of this complication are valuable. Several intraoperative test for checking bile leaks after hepatic resection have been described in literature and routinely used by surgical teams. One of particular efficiency, described for the first time by Nadalin et al 2008, is the so-called ‘white test’.7 With this procedure, 10–30 mL of sterile fat emulsion 5% is injected through the cystic duct while manually occluding the distal common bile duct. As the biliary tree fills with fat emulsion solution, leakage of the white fluid is visualised on the raw surface of the liver resection margin. The detected leakages are closed by means of single stiches. The use of a sterile fat emulsion, which is white because of its fatty content, allows the clear intraoperative identification of open bile ducts at the liver resection surface. Other than methylene blue, sterile lipidic solutions have the advantage not to stain the liver surface, as can be easily washed off and the test can be repeated if needed. In clinical practice, fat emulsions are normally used for parenteral nutrition.
Nadalin et al described in the publication of 20087 that the performance of the white test could intraoperatively identify additional potential bile leakage in 74% of the patients. No complications caused by the administration of the fat emulsion were seen in the study. Moreover, Li et al 2009 describe in a prospective cohort study in 137 patients undergoing a major liver resection a reduction of postoperative bile leakage from 22% in the control group to 5% in the White test group (p=0.004).8 Leelawat et al found 2012 in a self controlled study with 30 patients that a bile leak was demonstrated in 8 patients (26.7%) by the conventional method (injection of 10–20 mL isotonic sodium chloride solution in the biliary tract) and in 19 patients (63.3%) by the white test (injection of 10–20 mL of a 5% sterile fat emulsion in the biliary tract).9 A systematic review and meta-analysis published by Wang et al 2013 found that bile leakage test reduced postoperative bile leakage and did not increase incidence of complications. Fat emulsion was the best choice of solution for the test.10 A second meta-analysis published by Linke et al 2015 found that the use of the White Test led to a significant reduction of postoperative biliary leakage (OR 0.3 (95% CI 0.14 to 0.63), p=0.002) and led to a significant higher intraoperative detection of biliary leakage (OR 0.03 (95% CI 0.02 to 0.07), p<0.00001).11 A unique RCT was published by Liu et al12 comparing the White test with no test including both major and minor hepatectomies including a total of 107 patients (53 in the intervention group and 54 in the control one). The primary endpoint of the study, on which sample size was calculated, was the number of bile leakage sites detected intraoperatively on the liver resection surface with and without the white test (with an expected difference of 2.0). This end point is weaker than the postoperative bile leakage rate, which is clinically significant. In 2018 Naga et al published a prospective randomised (not controlled) study comparing the white test (50 patients) with conventional saline test (50 patients) in detecting intraoperative bile leakage in liver resection.13 To perform the white test, 10–20 mL of a 5% sterile fat emulsion (diluted SMOFlipid20%) was used. The postoperative course for all patients showed that 22 (44%) patients of the saline test group had bile leakage, but in the White test group, only four (8%) patients had leakage. Otherwise, there was no significant difference between both groups regarding other complications, especially postoperative liver affection recorded by liver decompensation in the form of deterioration of the liver synthetic functions to exclude any toxic effect of the white test on the residual liver.
Several studies and meta-analyses8–13 show the superiority of the white test when compared with no bile leakage test as well as with leakage test with saline solution. In all studies, no complications derived from the performance of the white test are described. Liu et al12 report that the fat emulsion is easily recognised and innocuous to the tissues as previously reported by McFadden et al14 who used it for intraoperative localisation of urinary leakage. An overview of the literature is presented in table 1. The level of evidence of the existing literature on the topic is medium to low (grade 2b) based on the definition of evidence levels provided by the Centre of Evidence in Oxford.15
Literature overview about the performance of the white test for biliary leakage after liver surgery
As to the best of our knowledge, up to date no good quality RCT regarding that question has been performed, we propose to conduct a multicentric RCT with primary endpoint the rate of postoperative bile leakage with and without the intraoperative performance of the white test.
Methods and analysis
Fat emulsion
As all existing studies describe the use of a sterile fat emulsion normally used for parenteral nutrition for the performance of the White test without giving the commercial name, with exception of the study by Nage et al 201813 where SMOFlipid20% is used, we decided to use the same fat emulsion.
Regarding the concentration of the solution, only 4 studies report this to be 5%,7–9 13 the remaining studies describe only a ‘sterile fat emulsion’. According to the majority of the published studies, we will use SMOFlipid20% diluted to a concentration of 5% similarly to the study by Naga et al.13 The dilution will be done by adding intraoperatively to the 100 mL SMOFlipid20% 300 mL isotonic saline solution in order to obtain 400 mL of a 5% fat emulsion.
Design
The BiLe-Trial is an investigator-initiated randomised controlled, parallel group, double-blinded (patients and care givers with exceptions of the operatory team), multicentre superiority trial investigating the effectiveness of the white test with lipidic solution (SMOFlipid 5%, Fresenius Kabi) in the prevention of postoperative bile leakage after resection of two or more liver segments. Eligible patients will be randomised equally to either application of the white test or standard intraoperative procedure to identify bile leakage (visual control with a white gauze).
Planned start date and duration of the study
Patients’ recruitment started in March 2021. Duration of the study is 3 year. If after 1 year less then 70 patients will be included by the actual four participating centres, one or more university centres will additionally be included in the study.
Patient and public involvement
As bile leakage is a main complication after liver resection, there is a general interest oft he patient in strategies which can reduce the rate of this complication. As the intervention is limited to a 5 min longer operative time through the once administration of SMOF lipis, patients perceived burden of this study is very low. Because of this, patients were not involved in design of the study and no patient advisers are present. By obtaining the consent for the operation, elucidating risks and complications of the surgical procedure, patients are informed about the trial. They are informed that the administration of diluted SMOF lipid does not have side effects according to the current literature. As they are blinded, they will not know to which group they are assisgned. Study results will not be disseminated tot he participants. In case of personal interest each study participant can contact the principal investigator of the hospital in which the operation was performed and ask about trial results after official closure of the database and analysis of the data.
Trial population
All adult patients with an indication for elective resection of at least two liver anatomic segments because of primary benign or malignant liver disease as well as metastatic disease from four regional Swiss centres (cantonal hospitals of Aarau, Lugano, St. Gallen and Lucerne) will be assessed for eligibility. All patients undergoing an anatomic resection of two or more liver segments, including major anatomical hepatectomies, left lateral lobectomies or any other anatomical resection of minimum two segments will be included. In each of the four regional hospitals, all operations will be performed by the same surgical team.
Inclusion criteria
Participants fulfilling all of the following inclusion criteria are eligible for the study:
Patients who will receive an anatomical resection of two or more anatomic liver segments for any reason with simultaneous cholecystectomy in elective setting.
Patients who will receive an anatomical resection of two or more anatomic liver segments for any reason and already had a cholecystectomy if intraoperatively the cystic stump can be identified and opened.
Ability of subject to understand character and individual consequences of the clinical trial.
Informed consent has been documented by signature.
Exclusion criteria
The presence of any one of the following exclusion criteria will lead to exclusion of the participant:
Previous cholecystectomy if intraoperatively it is not possible to identify the cystic stump (this should be recognised before the intraoperative randomisation takes place)
Intraoperative hepaticojejunostomy (as for grade A bile leakage, which do not need intervention, it would not be possible to discriminate if the leakage originates from the bilio-digestive anastomosis or from the liver surface). If intraoperatively a hepatico-jejunostomy is necessary, the patient will not be randomised.
After the exploration phase at the beginning of the operation, the planned operation cannot be performed. If the patient is not resectable, he/she will not be randomised.
Hypersensitivity to fish, egg, soybean or peanut protein or to any of the active ingredients or excipients.
Age under 18, immunosuppression and pregnancy.
Emergency liver resection because of traumatic liver rupture.
Enrolment of the investigator, his/her family members, employees and other dependent persons.
Randomisation
Patients will be intraoperative randomised by a study nurse using a centralised web-based tool (in SecuTrial) in a 1:1 ratio between white test and standard procedure as shown in figure 1. Permuted-block randomisation will be used to provide treatment allocation in equal proportions, with block sizes that will be subject to random variation (2, 4 and 6 patients). This will be concealed to all investigators involved in the trial.

Standard Protocol Items: Recommendations for Interventional Trials diagram.
Randomisation will be stratified by centre to balance differences in the surgical procedure and general treatment regimes, in order to detect possible center effects and prevent a center bias. The randomisation arm will then be communicated intraoperatively by a phone call to the operating surgeon.
Surgical procedure
The transection of hepatic parenchyma will be made either by monopolar dissection of the capsule and CUSA dissection with selective ligature or clipping of venous and portal ramifications of >3 mm diameter, or using endovascular staplers or with diathermy (Ultracision, LigaSure, Thunderbeat) with in all cases supplementary hemostasis by bipolar coagulation. The surgical technique will be chosen according to the surgical team preference and confidence within these three allowed categories. All resections will be performed under intraoperative ultrasound control. A retrograde cholecystectomy will be performed if indicated according to the surgical procedure. If a cholecystectomy is not necessary, like in a segment 2/3 resection, this will be performed only if the patient is randomised to the intervention group in order to get access to the main bile duct via cystic stump. If cholecystectomy has already taken place in a previous operation, the cystic stump will be explored and if this could be easily reopened according to the experience of the operating surgeon the patient will be randomised, otherwise not. If the patient will be randomised to the intervention group, the cystic stump will be reopened.
At the end of resection, the patient will be then intraoperatively randomised through a phone call to a study nurse working at the surgical department of the hospital of St. Gallen (KSSG) who has access to SecuTrial, the Clinical Data Management System used for data collection, to either the intervention (identification of open bile ducts with the white test) or the control group (identification of open bile ducts with a white gauze on the liver resection surface in addition to the visual control). At the end of the operation, a drainage will be placed at the liver resection surface in all patients regardless the randomisation group. The type of drainage (active or passive) is chosen according to the internal standard of each clinic. An additional intrabdominal fixation beside the standard fixation to the skin of the drainage can also be performed according to the surgeon’s preference. At the third postoperative day, the drainage is monitored for any biliary content in the fluid (measurement of bilirubin in the drain fluid or macroscopic presence of bile). The aim of this choice is to get uniformity in all surgical teams and keep blinding.
Intervention: ‘white test group’
If a patient is randomised to the intervention arm, open bile ducts are identified by administration of a sterile lipidic solution retrogradely through the cystic duct in addition to visual control of the resected area.
In particular, 20–40 mL SMOFlipid 5% will be injected in the cystic stump, directing the flow to the intrahepatic ducts by clamping the distal part of the common bile duct manually. If the quantity of lipidic solution is not enough, more saline solution will be injected. The resected area will be inspected carefully for any white liquid leakage. If a leak is found, a direct suture will be performed. After closing the leakage site, the residual fat emulsion on the resection surface (potentially masking other bile leakages) will be washed off and the white test is repeated to detect additional bile leakages. The test will be repeated if the leak site is not clearly recognised and for final check after the leak sutures. At the end of the white test, residual fat emulsion is washed out from the biliary tract by a low pressure infusion of 20 to 50 mL of saline solution, as described by Nadalin et al 2008,7 in order to avoid any significant enteral absorption.
Control: ‘white gauze group’
In the control arm, open bile ducts will be identified with a white gauze on the liver resection surface in addition to the visual control. If a leak is found, a direct suture will be performed and the test with white gauze repeated.
Some variation of the surgical technique (eg, type of drainage, intra-abdominal fixation of the drainage, device used for parenchymal dissection) at the discretion of the operating surgeon are allowed.
Patient blinding
In order to achieve blinding two different study teams are present in each clinic: a ‘masked’ and an ‘unmasked’ team. To the unmasked team belong all surgeons performing the operation and the masked team consists of data collectors and outcome assessors. Documents are also physically stored separately in a masked and unmasked folder. The unmasked folder is stored by the operating surgeons and contains information about randomisation group, batch number of SMOF lipid and NaCl which have also to be recorded. In the responsibility log stored in the Investigator Site File (both masked and unmasked), names of colleagues involved in the study are listed and it is clearly defined to which team each person belongs. For sure the surgeons who perform the operation will also visit the patients during the daily regular round but they do no’t have access to SecuTrial and consequently they cannot entry data. They are only involved in performing the operation. In the operating report is written ‘the patient participates to the BiLe trial. He/she was intraoperatively randomized and treated accordingly’. Both team members can screen patients and obtain the patient informed consent. The trial statistician is also not blinded, he/she will get access to the data only after closure of the study. If necessary, data of a patient can be unblinded at any time by the sponsor. The unblinding is permanent for this specific patient.
Primary outcome
Comparison of the rate of postoperative bile leakage within 30 postoperative days in both groups (as a binary endpoint, yes/no)
Definition of bile leakage: according to the definition by the International Study Group of Liver Surgery,16 a bile leakage was defined as bilirubin concentration in the drain fluid at least three times the serum bilirubin concentration on or after postoperative day 3 or as the need for radiologic or operative intervention resulting from biliary collections or bile peritonitis. The severity of bile leakage was classified according to its impact on patients’ clinical management. Grade A bile leakage causes no change in patients’ clinical management. Grade B bile leakage requires active therapeutic intervention but is manageable without relaparotomy, whereas in grade C bile leakage relaparotomy is required.
Secondary outcomes
Severity of bile leakage (grades A, B or C according to the definition by Koch et al).16
In-hospital Mortality and Morbidity other than related to the bile leakage as for example pneumonia, wound infection and wound dehiscence after laparotomy, need for any radiological examination without intervention, reoperation (according to the Clavien Dindo Classification).17
ERCP (yes/no).
Percutaneous transhepatic biliary drainage (yes/no).
Interventional drainage (yes/no).
Reoperation (yes/no).
Intensive care unit stay (in days).
Total hospital stay (in days).
Other outcomes
Bile leakage rate and severity in each group according to the device used to transect the liver surface.
Bile leakage rate and severity in each group according to the presence or absence of liver cirrhosis/steatosis.
Total operation time (in min) and parenchymal dissection time (in min).
Intraoperative blood loss according to the device used for parenchymal transection.
As several techniques exist to transect the liver parenchyma, we will perform subgroup analyses analysing the bile leakage rate and the severity according to the device used as well as according to the presence or absence of liver cirrhosis/fibrosis. In the four participating centres, three categories of instruments are used: diathermy (Ultracision, LigaSure, Thunderbeat), vascular stapler and CUSA according to the preference of the surgeon. The time needed for the dissection as well as the total operation time and blood loss will be compared according to the device used for the parenchymal transection regardless the administration of SMOFlipid.
Data collection and patient follow-up
All data will be collected via SecuTrial (electronic case report form eCRF). The database was set by the data management team of the clinical trial unit of Basel, Switzerland. In particular, baseline data on age, sex, type of resection, previous cholecystectomy, preoperative diagnosis, indication for surgery as well as intraoperative variables (blood loss, operation time, device used to transect the liver parenchyma, transection time, type of liver resection and technique used—open or laparoscopic), histology and postoperative complications will be recorded. All patients, regardless the allocation group (white test or control) will have standard postoperative care, related to the clinical evolution. Bilirubin measurement in the drain fluid on postoperative day 3 will be performed as routinely. All serious adverse events (SAEs) are recorded in SecuTrial and reported within 24 hours to the sponsor. The sponsor submits annually the Annual Safety Report (ASR) to the competent authorities.
No supplementary exams or outpatients visits are planned specifically to the study. Study duration for each patient will be 30 days (±7 days) from the day of randomisation. At day 30th (±7 days), the patient will be routinely seen in the surgical consultation hour or at the daily round if still in hospital. If the patient is already discharged from the hospital and the surgical consultation hour is planned outside the time 30th (±7 days), a phone call in the rehabilitation clinic or to the patient itself will be performed. All patients will follow the standard follow-up according to clinical status and local hepatobiliarypancreatic teams practice.
The total duration of the study is set to 3 years; the monitoring will be performed by the monitoring team of the Clinical Trial Unit of the University Hospital of Basel, Switzerland.
Statistical aspects
Sample size calculation
The BiLe-Trial is designed as a superiority trial, hypothesising that the intraoperative performance of the white test with lipidic solution reduces the rate of postoperative bile leakage compared with the use of a white gauze applied on the resected liver surface.
The sample size was determined via simulations of the primary analysis. The simulations were based on the assumption that the intraoperative use of the white test with SMOFlipid 5% will lead to a postoperative reduction of the bile leakage rate from 21% in the control group (Nadalin et al and our retrospective data) to 7% in the intervention group. To detect this difference with a statistical power of 80% using Pearson’s χ2 test (with continuity correction) at a two-sided 0.05 significance level a sample size of 105 patients was required for each group, which means that 210 patients should be included in the study. No interim analysis is planned. The assumptions about the bile leakage rates were derived from evidence in the literature in conjunction with experience at hospitals participating in the study and currently using one of the studied treatments. The Cantonal Hospital Aarau is currently using the control treatment and saw a bile leakage rate of 19% among patients undergoing liver resection of two or three segments during the past years. A prospective cohort study of patients undergoing major (ie, at least three segments) liver resection found a bile leakage rate of 22.9% for the control group.8 The same cohort study found a bile leakage of 5.3% for the treatment group and also another observational study found a similar bile leakage rate of 5.1% among patients undergoing major liver resection surgery with lipidic solution.7 By contrast, the Cantonal Hospital of St. Gallen is routinely using lipidic solution in addition to standard methods and estimated their bile leakage rate at 10%. Hence, for both study groups, we assume a bile leakage rate that lies in between those observed.
Statistical analysis
The primary analysis investigates whether the bile leakage rate (within 30 days) among patients with SMOFlipid 5%-based detection of open bile ducts is different from the rate in the control group, using a Pearson’s χ2 test (with continuity correction) at significance level α=0.05.
The secondary endpoints will be analysed using logistic regression models for the binary ones and suitable generalised regression models for the remaining endpoints. Since there might be some heterogeneity between the participating centres, we will estimate each regression model two times: once ignoring center-specific heterogeneity and once adding a random effect for the centre variable in the model. A secondary analysis of the primary endpoint will use a logistic regression model with a center-specific random effect. All results of the secondary analyses will be reported as estimated effects and their 95% confidence intervals. No correction for multiplicity will be made.
The Full Analysis Set (FAS) will include all study participants who were randomised. The Per Protocol Set (PPS) will include all patients in the FAS for whom the treatment and follow-up were completed as planned in the study protocol. All statistical analyses will be performed on the FAS according to the intention-to-treat principle. In addition, the primary analysis and selected secondary analyses will be repeated using the PPS instead to assess robustness of the findings.
Missing primary and secondary endpoints will be imputed for the FAS analyses. If less than 5% of the primary endpoint data is missing, the few missing cases will be imputed two times: once with the best case (no leakage) and once with the worst case (leakage). If more than 5% of data is missing, multiple imputation schemes will be considered. In addition, complete cases analyses will be performed to evaluate sensitivity of the results to different assumptions about the unobserved data.
Ethics and dissemination
Ethical and safety considerations
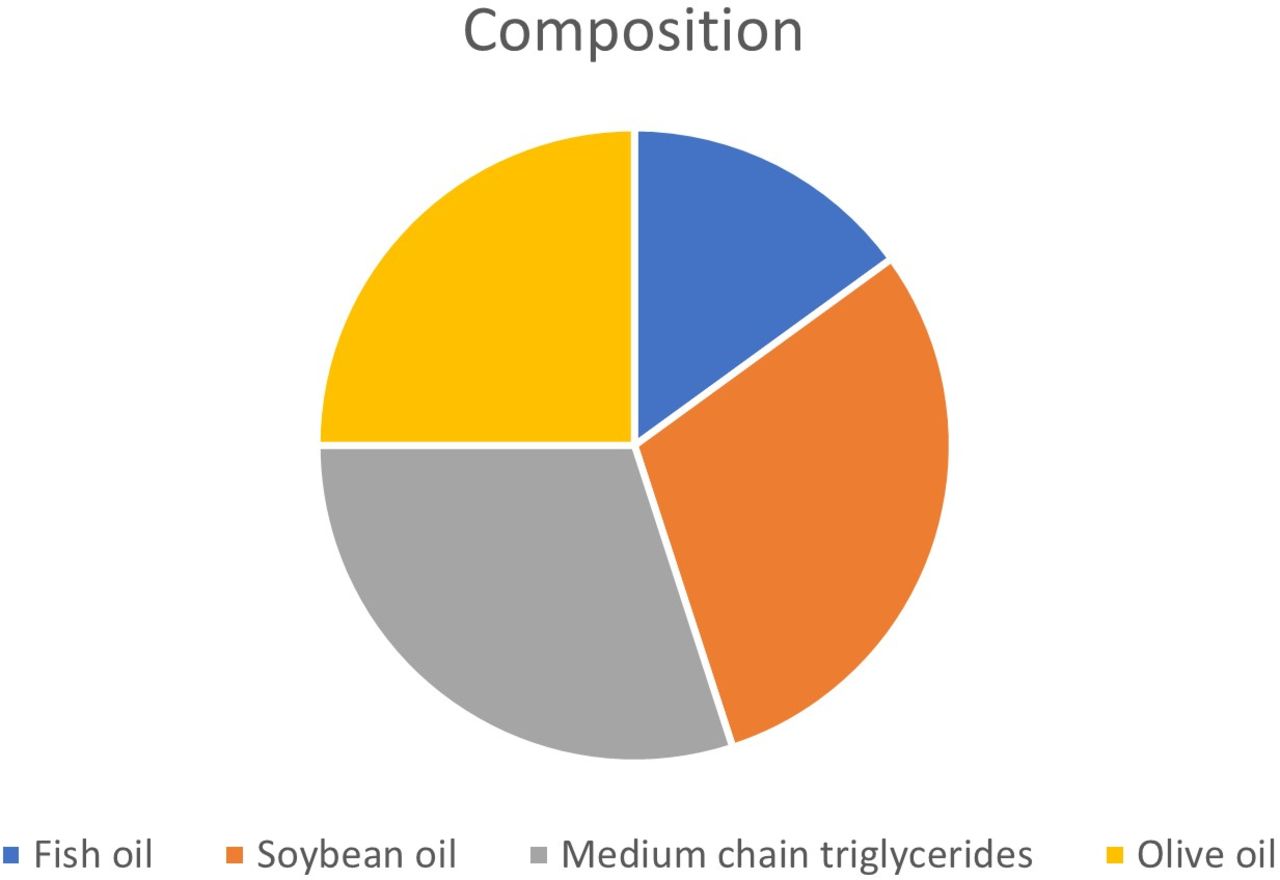
This study will be conducted in compliance with the protocol, the current version of the Declaration of Helsinki,18 the ICH-GCP (International Conference for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use - Good Clinical Practice) guidelines19 20 as well as all national legal and regulatory requirements. Swissethics, after approvation of the single cantonal ethic committee (ethic committee nord west and central Switzerland (EKNZ), ethic committee of east Switzerland (EKOS) and ethic committee of Ticino (TI)) approved the study (BASEC ID 2020-02081). As our study is a clinical trial on medicinal product classified as risk category B, the additional approbation by Swissmedic (ID 2020DR3177) was obtained. The approbation by Swissmedic, the Swiss Agency for therapeutic Products was necessary as SMOFlipid will be administered in a way (biliary system) other than the one for that the approval exists (intravenous) as well as for a new indication (prevention of bile leakage instead of parenteral nutrition). Preclinical studies on animals have not demonstrated other adverse events than those described for lipidic solution’s intravenous overdose. Concerning local tolerance, this has been studied on rabbits showing a slight rapidly reversible local inflammation after intra-arterial, extravenous and subcutaneous administration. A moderate rapidly reversible local inflammation is described for intramuscular administration in some animals. As SMOFlipid consists of a mixture of oils (figure 2), side effects resulting from the once intraoperative application of 20–40 mL in the biliary system with consecutive enteral absorption are not expected. An intraperitoneal spread of the substance is not expected as a suction system is used during the operation. As described in the introduction, several studies showed no side effects deriving by the administration of sterile fat emulsions through the biliary tree. Additionally, in order to administer the lower possible doses, as described by the majority of the authors using the white test, SMOFlipid20% will be diluted to a solution of 5% of concentration by adding 300 mL NaCl to 100 mL SMOFlipid20%.

{kind=link}
{kind=link}
Composition of SMOFlipid 20% according to the information sheet (https://www.fresenius-kabi.com/en-ca/products/lipid-emulsions).
All SAE will be collected in the eCRF and reported from the principal investigator to the sponsor. The sponsor is responsible for the compilation of the ASR. SAE resulting in death will be communicated within 24 hours from the sponsor to the ethic committee.
Dissemination policy
The results of the BiLe-Trial will be submitted to a peer-reviewed journal regardless of the outcome. Authorship will be based on international guidelines.
Ethics statements
References
Footnotes
Contributors GM, AC, IT and MK contributed substantial to conception and design of the study. AW wrote the statistical analysis plan and performed sample size calculation. AW, AS, JM, BMS, PM-J and MH revised all documents for authorities and had continuously an advisory role in planning the study. All authors gave the final approval of the version to be published.
Funding The study will be supported by the Research Council of the cantonal hospital of Aarau (KSA) and by the Gottfried and Julia Bangerter-Rhyner-Stiftung.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.