Article Text

Abstract
Objective The dural sealant patch (DSP) is designed for watertight dural closure after cranial surgery. The goal of this study is to assess, for the first time, safety and performance of the DSP as a means of reducing cerebrospinal fluid (CSF) leakage in patients undergoing elective cranial intradural surgery with a dural closure procedure.
Design First in human, open-label, single-arm, multicentre study with 360-day (12 months) follow-up.
Setting Three large tertiary reference neurosurgical centres, two in the Netherlands and one in Switzerland.
Participants Forty patients undergoing elective cranial neurosurgical procedures, stratified into 34 supratentorial and six infratentorial trepanations.
Intervention Each patient received one DSP after cranial surgery and closure of the dura mater with sutures.
Outcome measures Primary composite endpoint was occurrence of one of the following events: postoperative percutaneous CSF leakage, intraoperative leakage at 20 cm H2O positive end-expiratory pressure or postoperative wound infection. Overall success was defined as achieving the primary endpoint in no more than two patients. Secondary endpoints were device-related serious adverse events or adverse events (AEs), pseudomeningocele and thickness of dura+DSP. Additional endpoints were reoperation in 30 days and user satisfaction.
Results No patients met the primary endpoint. No device-related (serious) AEs were observed. There were two incidences of self-limiting pseudomeningocele as confirmed on MRI. Thickness of dura and DSP were (mean±SD) 3.5 mm±2.0 at day 7 and 2.1 mm±1.2 at day 90. No patients were reoperated within 30 days. Users reported a satisfactory design and intuitive application.
Conclusions DSP, later officially named Liqoseal, is a safe and potentially efficacious device for reducing CSF leakage after intracranial surgery, with favourable clinical handling characteristics. A randomised controlled trial is needed to assess Liqoseal efficacy against the best current practice for reducing postoperative CSF leakage.
Trial registration number NCT03566602.
- neurosurgery
- clinical trials
Data availability statement
Data are available in a public, open-access repository. Extra data can be accessed via the Dryad data repository at https://datadryad.org/stash/dataset/doi:10.5061/dryad.4j0zpc8br.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The trial studies a device to prevent postoperative cerebrospinal fluid leakage, which is one of the most common neurosurgical complications.
The study protocol was performed in multiple centres, registered, prepublished and strictly followed.
The composite endpoint of the trial reduced the number of inclusions needed.
The study did not involve a comparison to current clinical standard and has a potential selection bias, so generalisation of results with regard to DSP efficacy needs to be cautiously undertaken.
Introduction
Cerebrospinal fluid (CSF) leakage is one of the most common neurosurgical complications, occurring approximately in 8% of surgical cases with a higher incidence in complicated skull base surgery, intradural spine surgery and surgery of the posterior fossa.1–3 Most patients with CSF leakage require a prolonged hospital stay, antibiotic treatment for meningitis, external lumbar drainage, reoperation or a combination of these measures. CSF leakage leads to significant patient burden and expense, with an estimated cost of US$10 000–15 000 per patient per leakage.2 The use of a dural sealant as an adjunct to primary dural closure is often assumed to further prevent CSF leakage. However, initial approval for liquid sealant was based only on successful intraoperative performance, rates of CSF leakage and other clinically relevant postoperative outcomes, which were similar compared with controls.3–5
The sponsor of this study (Polyganics BV, Groningen, the Netherlands) has developed, in close cooperation with our research group, a dural sealant patch (DSP) (figure 1). This bioresorbable patch is intended for use as an adjunct to standard methods of dural closure, such as suturing, to provide a watertight closure of the dura mater to prevent CSF leakage after dural closure procedure. It supports immediate watertight bonding to dura without a liquid component or spray.

Dural sealant patch/Liqoseal. Produced by Polyganics BV, Groningen, the Netherlands.
Preclinical studies showed better adherence to dura and higher burst pressures than currently used sealants. Biological safety hazards of DSP have been addressed according to International Organization for Standardization (ISO) guideline 10993 (biological evaluation of medical devices)6 in a series of in vitro and/or in vivo studies: cytotoxicity; sensitisation; irritation; acute, subacute and subchronic toxicity; pyrogenicity; hemocompatibility; genotoxicity; neurotoxicity; local effects; and in vivo degradation up to 12 months. A large implant study in a porcine model showed no arachnoidal adherence or reaction of the brain when directly in contact with the brain (submitted). Based on these data, DSP was considered safe for implantation.
Until the current study, DSP was not tested in human subjects yet. This study aims to study clinical safety and performance of the DSP in reducing CSF leakage in patients undergoing elective cranial intradural surgery with dural closure.
Methods
This study was conducted as an open-label, single-arm, multicentre study. The study was performed in accordance with the Medical Device Directive (93/42/EEC and Meical Devices Document (MEDDEV) 2.7/3 rev. 3, 2015,7 MEDDEV 2.7/4,8 World Medical Association Declaration of Helsinki9 and ISO 14155:2011.10 The ENCASE protocol (supplementary material: Clinical Investigational Plan ENCASE) was approved by the Medical Ethical Commission in Utrecht, the Netherlands (NL64477.041.18), the Dutch Inspection for Healthcare and Youth (IGJ) and the Swiss Medical Ethical Board (BASEC 2018–01073). The protocol has been previously published open access in detail11 (online supplemental appendix 1). The study coordinator and investigators followed accredited Good Clinical Practice (GCP) training, and the study was performed according to GCP regulations. We used the ‘Reporting Guidelines Checklist for IDEAL Stage 4’ in writing our manuscript.12
Supplemental material
Public involvement
Patients or the public were not involved in the design, conduct or reporting of our research. The study results were disseminated to study participants via email.
Setting
Three large, tertiary reference neurosurgical centres, two in the Netherlands and one in Switzerland.
Patients
Forty adult patients scheduled for elective cranial surgery with a dural opening of minimal 2 cm were enrolled for this study. At the three individual study centres, patients were screened for participation. Patients needing an intradural drain, electrodes or other devices passing the dura mater after surgery were excluded. All patients gave written consent. Alternatives were discussed, and patients were specifically informed that this was the first clinical application of this device. We stratified into 34 supratentorial and six infratentorial trepanations. First enrolment was on 11 October 2018, last enrolment on 30 April 2019 and last follow-up on 29 April 2020. Detailed inclusion criteria have been published previously.11 Baseline characteristics are listed in table 1.
Baseline
DSP
DSP (figure 1) is a flexible patch and consists of two layers: the adhesive layer (white) and the sealing layer (blue). The blue layer consists of biodegradable polyesterurethane (PU). The white adhesive layer is foam-shaped and consists of bioresorbable copolyester. The white foam covalently bonds to the dura due to the incorporated N-hydroxylsuccinimide functionalized polyethylene glycol (PEG-NHS) adhesive component and buffer salt. This layer reacts with amines in the dural tissue in a moist environment, forming covalent bonds between the device and the tissue.
Procedure
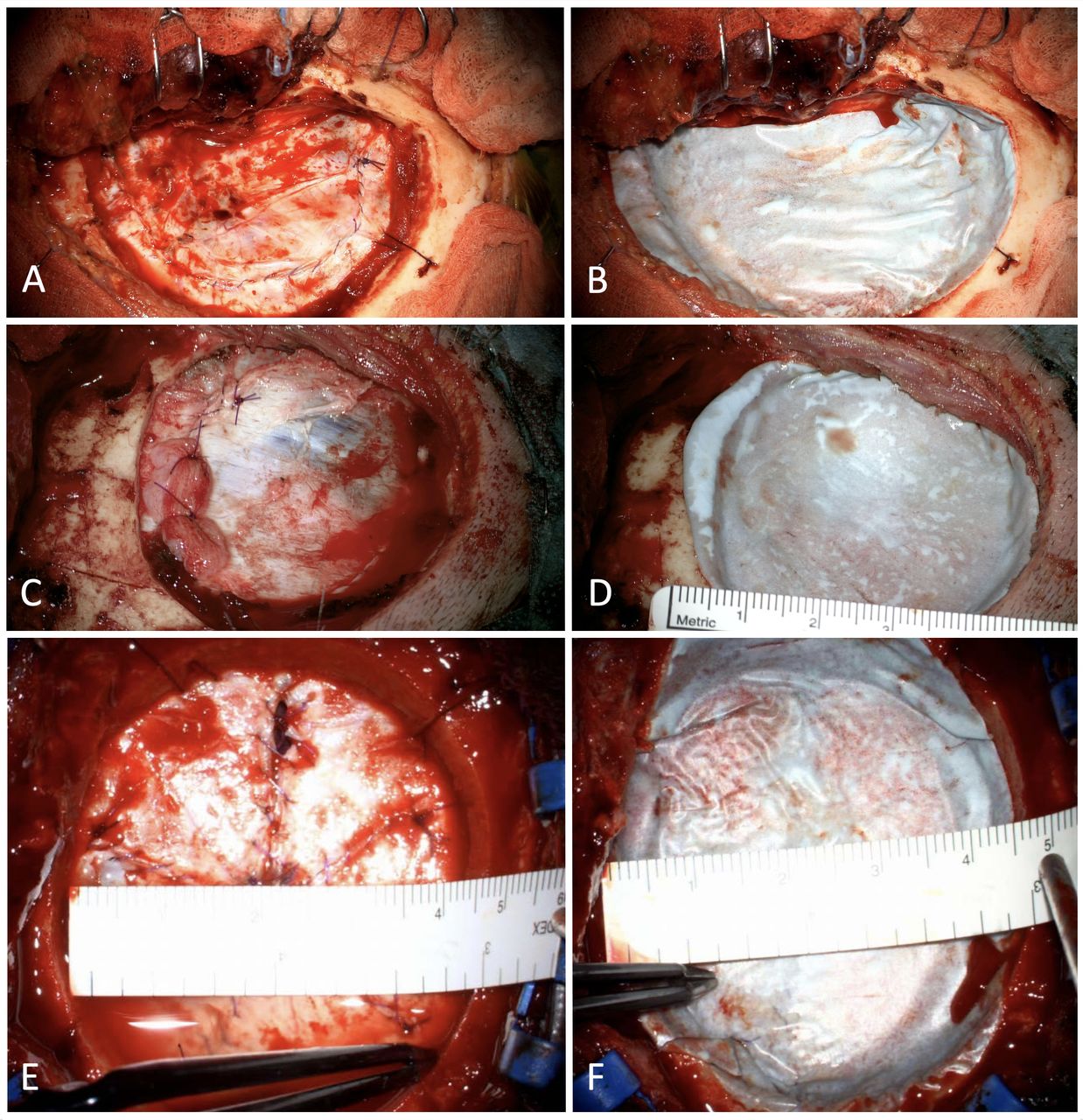
Minimally two surgeons per centre participated in the trial; all were individually trained on the protocol. Before dura mature closure, the positive end-expiratory pressure (PEEP) was increased to 20 cm H2O for 20 s to check for haemostasis video 1. The dura mater was then closed by suturing with the intention for watertight closure. However, a maximal dural gap of 3 mm was accepted (figure 2A). A substitute (autologous tissue only) could be used by the discretion of the surgeon (figure 2C). The PEEP was increased for the second time to 20 cm H2O for 20 s to verify saline or CSF leakage out of the dural closure (figure 2E). Each patient then received one DSP after closure of the dura mater. The patch had to overlap the dural opening for at least 5 mm and was slightly compressed with a moist gauze for 2 min (figure 2B, D and F). Exactly 2 min after finishing compression, the PEEP was increased to 20 cm H2O for 20 s for the third time. The surgeon assessed CSF leakage during and after this PEEP increase until skin closure. All procedures were filmed (video 1) and stored on file.
{kind=link}
{kind=link}
Three patients before and after application of dural sealant patch (DSP). (A) and (B) Patient 6; (C) and (D) patient 14, a piece of muscle as dural substitute is used; and (E) and (F) patient 30, the saline leak is seen basal at 20 cm H2O before DSP application.
Follow-up
Follow-up of the subjects was performed clinically at day 7 (or at discharge, whichever came first) and at 30, 90 and 360 days after implantation. Additionally, subjects underwent an MRI on day 7 or discharge (whichever came first) and on day 90. All imaging was evaluated and scored by an independent neuroradiologist. The study was controlled and monitored by a clinical research organisation (CRO), Genae (Antwerpen, Belgium).
Endpoints
Primary endpoints
Primary composite endpoint was defined as the occurrence of one of the following events:
Incidence of wound infection within 30 days as defined in accordance with Centers for Disease Control and Prevention guidelines for superficial incisional, deep incisional and organ space infections (safety endpoint).
Incidence of intraoperative CSF leakage after patch application at 20 cm H2O of PEEP (efficacy endpoint).
Incidence of percutaneous CSF leak confirmed by β-2 transferrin test up to 30 days after surgery (efficacy endpoint).
Secondary endpoints
Incidence of device-related serious adverse event (SAEs) and adverse events (AEs) throughout the study up to 360 days after surgery. (safety endpoint).
Incidence of wound infections up to 90 days after surgery (safety and efficacy endpoint).
Incidence of percutaneous CSF leak up to 90 days after surgery (efficacy endpoint).
Incidence of pseudomeningocele with the need of puncture, external lumbar drainage or surgical evacuation as assessed by treating physician up to 90 days after surgery.
Incidence of pseudomeningocele >20 cc as confirmed on MRI (efficacy endpoint).
Thickness dura mater and DSP (combined) in millimetre analysed with MRI (safety endpoint).
Additional endpoints
Incidence of complication requiring a reintervention up to 30 days after surgery. (safety endpoint).
Ease of use and application of the DSP (closed-end questionnaire) (online supplemental appendix 2).
Supplemental material
Statistics
The primary (composite) endpoint was scored ‘yes’ if any of the primary outcome events occurred and ‘no’ otherwise. This binary outcome was assumed to follow a binomial distribution. Overall study success was defined as the proportion meeting the primary endpoint in 7% or less in the study population, based on previously reported complication rates.1 2 4 13 Therefore, the number of patients experiencing the primary outcome measure would have to be no more than two for study success. The sample size calculation was based on using a CI approach for one proportion (exact Clopper-Pearson). Based on an expected proportion of 7% on scoring ‘yes’ on the primary composite endpoint and a target width of 0.20, a 95% CI of 0.012 to 0.209 is obtained with a sample size of 35. Allowing for 12.5% dropout, we aimed to recruit 40 patients for this study.
Data and safety monitoring
Details on data management and safety were published before.11 Monitoring was provided by a professional independent CRO (Genae, Antwerp, Belgium). The monitor verified all critical data points against the source documents and issued electronic queries for the authorised clinical site personnel to respond. A critical quality control was performed for the first two subjects at each site. A full quality control was performed on the monitored data throughout the clinical investigation, and queries were issued where needed. This process was repeated until the end of the clinical investigation so as to allow for a timeline freezing of the database for statistical analysis.
An independent Data Safety Monitoring Board (DSMB) was installed, consisting of three neurosurgeons not participating in the study with no competing interests, assisted by an independent statistician (online supplemental appendix 3: DSMB charter). The DSMB reviewed all data relating to safety and performance and had a final say on study continuation, thereby ensuring the safety, scientific validity and merit of the study. DSMB analysis was performed after five patients accomplished 30-day follow-up and after 10 patients accomplished 30-day follow-up, at study enrolment completion, at 90-day follow-up completion and at 360-day follow-up completion. At the end of the study, all investigators had access to the final dataset.
Supplemental material
Results
We screened 46 patients and included 40 patients; four patients failed screening criteria, and two patients withdrew before application. Of the 40 included patients, 24 patients were women. Thirty-four patients received a supratentorial DSP application and six patients an infratentorial DSP application (table 1).
Primary endpoints
No patient reached a primary safety or efficacy endpoint, and therefore, the primary composite endpoint was not reached in any patient (table 2).
Outcome
Secondary endpoints
During the 360-day follow-up, 214 total AEs were reported. Of these, 18 AEs were reported to be SAEs in six subjects (online supplemental appendix 4). None of the AEs were judged ‘definitive device related’ by the study coordinator nor by the DSMB. One of the SAEs was marked with ‘possibly device related’. This subject was diagnosed with a chemical meningitis, after craniotomy for craniopharyngioma. The direct relation with the study device seems questionable; however, a potential relationship could not be ruled out. The other recorded (serious) AEs were not related to the device.
Supplemental material
No wound infection or percutaneous CSF leak was diagnosed during 90 days of follow-up.
Two subjects reached the secondary efficacy endpoint of a pseudomeningocele of >20 cc confirmed by MRI. These were both self-limiting and proved to be resorbed at 90 days by MRI. These pseudomeningoceles had no clinical consequences for the patients.
Thickness measurements showed no clinically significant swelling of the DSP. Compared with the device thickness before application (~5 mm), the mean thickness after application did not exceed this specified thickness. At day 7, a mean thickness of 3.5 mm (SD 2.0) was measured, and at 3 months, a thickness of 2.1 mm (SD 1.2). In 65% of the subjects, the device was still separately visible on MRI at day 7, which decreased to 20% by day 90.
Additional endpoints
No patient underwent a reoperation within 30 days after surgery.
After every procedure, the neurosurgeon who applied the device answered ‘good ‘or ‘excellent’ on the question ‘how intuitive was the application of the device?’. Detailed user experience is stated in online supplemental appendix 2.
DSMB evaluation
The final evaluation performed by the DSMB up to day 360 after the last implantation resulted in a recommendation to terminate the trial without any safety concerns. Based on the interim results of the current study combined with all preclinical date CE certification was granted to the DSP on 7 January 2020, which was renamed ‘Liqoseal’.
Discussion
With this first clinical study of the DSP (Polyganics BV, Groningen), we demonstrate its general safety and potential efficacy in elective cranial surgery, with none of the patients reaching a primary safety or efficacy endpoint.
The strengths of the current study are a prepublished protocol, a strict adherence to study procedures by training a selective group of surgeons, the involvement of a CRO and its multicentre organisation. Thereby, the use of a composite endpoint reduced sample size.
However, the current study has also some weaknesses. First, a randomised controlled trial (RCT) investigating the safety and efficacy might have provided more robust data regarding the success of DSP. The current trial was primarily a safety trial with a minimal number of patients using a composite endpoint and using a reference rate of published complications to show an effect. We chose this design because a direct RCT was regarded as an unacceptable ethical and financial risk.
A second potential weakness of this study is that one of the primary outcome measures (incidence of intraoperative CSF leakage) was assessed by the operating surgeon, which could have theoretically introduced misclassification of patients and therefore have positively influenced the primary outcome. To prevent this, all procedures had to be filmed and saved in the study database.
Finally, the current study harbours a selective patient population, because we tried to make the ENCASE study population as uniform as possible. Since biocompatibility of autologous tissue is uniform and well described,14 only this was allowed as a substitute. However, therefore, the interaction with other artificial substitutes remains unknown. Trauma, endoscopic surgeries and spinal surgeries with dural opening were also excluded, while these indications are associated with a higher CSF leakage risk. The added value of DSP in the excluded indications is potentially large but still has to be evaluated more in detail.
Closing the supratentorial dura with or without sealant and its role in CSF leak prevention are the subject of an ongoing debate. Kinaci et al5 performed a meta-analysis of 2321 intradural cranial cases showing no significant difference in CSF leakage rate between the use of a dural sealant (8.2%) and primary closure only (8.4%). Significant difference was found regarding surgical site infection, which was less seen in cases with sealants (RR 0.25, CI 0.13 to 0.48). Osbun et al10 performed a large RCT comparing dural sealing with a PEG hydrogel with ‘standard of care’. The absence of CSF leakage at intraoperative Valsalva manoeuvre was used as an inclusion criterium, not as a result variable. In total, 30% was infratentorial and 70% supratentorial, comparable with the current study. Unplanned reintervention rate was 4.2% (study group) versus 4.3% (control), surgical wound complications 3.3% versus 4.3% and postoperative CSF leak 0.8% versus 1.7%. Hutter et al1 performed an RCT comparing standard dural closure using suturing alone with the addition of TachoSil on top. In total, 19% of the procedures were infratentorial and 81% supratentorial. The authors regarded >20 cc pseudomeningocele an indication for treatment, which was also defined as CSF leakage. The difference in leakage rate was not significant with 9.7% in the TachoSil and 17.2% in the control group. Wound infection was 0.9% versus 4.3%. Although these studies are not fully comparable with the current study, we seem to show beneficial results in the current study with neither CSF leakage nor infections and 5% pseudomeningocele >20 cc (which were self-limiting).
Based on the current study, the DSP was CE certified and renamed ‘Liqoseal’. To rigorously assess Liqoseal efficacy against the best current practice for reducing postoperative CSF leakage, we have designed a subsequent RCT (ENCASE II, registered on ClinicalTrials.gov under NCT04086550). In this trial, only posterior fossa patients will be included, which are at higher risk for postoperative CSF leak than supratentorial patients. Clinically meaningful outcomes will be compared between Liqoseal and current standard practice. This study is named ENCASE II and is planned to start recruitment Q2 2021.
In conclusion, DSP/Liqoseal is a safe and potentially efficacious device for reducing CSF leakage after intracranial surgery with favourable clinical handling characteristics.
Data availability statement
Data are available in a public, open-access repository. Extra data can be accessed via the Dryad data repository at https://datadryad.org/stash/dataset/doi:10.5061/dryad.4j0zpc8br.
Ethics statements
Acknowledgments
We want to thank Ester Maas-Soer for her assistance in study design and coordination. We want to thank Berber Zweedijk, Paula van Limpt and Elisabeth Jehli for their indispensable help.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors TVD designed the study with Esther Maas-Soer (on behalf of the sponsor) and JWD. TVD, MRG, MS, BB and JF included and operated the patients. TVD, MRG, MS, BB, JWD, JF, PD acquired data assisted by Berber Zweedijk, Paula van Limpt and Elisabeth Jehli. TVD, MRG, and CRO (Genae, Antwerp, Belgium) analysed the data. CRO checked the data and provided statistical advice with MRG. TVD wrote the paper assisted by MRG, BB and AC. MS, BB, AC, JWD, JF, PD, PR and LPR corrected the manuscript versions. All authors approved the manuscript version for publication.
Funding This study was funded by Polyganics BV, Rozenburglaan 15A, 9727 DL Groningen, The Netherlands. Grant number: N/A.
Competing interests First author received a consultancy fee in the design phase of the product from Polyganics BV, the Netherlands. None of the authors have any other financial interest in the product or Polyganics BV in general.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.