Article Text

Abstract
Introduction Respiratory distress syndrome is a complication of prematurity and extremely preterm infants born before 28 weeks’ gestation often require endotracheal intubation and mechanical ventilation. In this high-risk population, mechanical ventilation is associated with lung injury and contributes to bronchopulmonary dysplasia. Therefore, clinicians attempt to extubate infants as quickly and use non-invasive respiratory support such as nasal continuous positive airway pressure (CPAP) to facilitate the transition. However, approximately 60% of extremely preterm infants experience ‘extubation failure’ and require reintubation. While CPAP pressures of 5–8 cm H2O are commonly used, the optimal CPAP pressure is unknown, and higher pressures may be beneficial in avoiding extubation failure. Our trial is the Extubation CPAP Level Assessment Trial (ÉCLAT). The aim of this trial is to compare higher CPAP pressures 9–11 cm H2O with a current standard pressures of 6–8 cmH2O on extubation failure in extremely preterm infants.
Methods and analysis 200 extremely preterm infants will be recruited prior to their first extubation from mechanical ventilation to CPAP. This is a parallel group randomised controlled trial. Infants will be randomised to one of two set CPAP pressures: CPAP 10 cmH2O (intervention) or CPAP 7 cmH2O (control). The primary outcome will be extubation failure (reintubation) within 7 days. Statistical analysis will follow standard methods for randomised trials on an intention to treat basis. For the primary outcome, this will be by intention to treat, adjusted for the prerandomisation strata (GA and centre). We will use the appropriate parametric and non-parametric statistical tests.
Ethics and dissemination Ethics approval has been granted by the Monash Health Human Research Ethics Committees. Amendments to the trial protocol will be submitted for approval. The findings of this study will be written into a clinical trial report manuscript and disseminated via peer-reviewed journals (on-line or in press) and presented at national and international conferences.
Trial registration number
ACTRN12618001638224; pre-results.
- Extremely preterm
- infant
- endotracheal extubation
- continuous positive airway pressure
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study is a multicentre randomised controlled trial.
This study is the largest trial comparing continuous positive airway pressure (CPAP) pressure ranges to reduce extubation failure in extremely preterm infants.
CPAP pressures as high as 9–11 cm H2O are yet to be formally evaluated in extremely preterm infants.
Due to the nature of the interventions blinding of patients and clinicians are unable to occur.
Introduction
Respiratory distress syndrome (RDS) is common in preterm infants, and almost universal in extremely preterm infants born <28 weeks’ gestation. In this high-risk population, bronchopulmonary dysplasia (BPD), the chronic lung disease of prematurity, is a major morbidity following RDS and its treatment.1 Many extremely preterm infants require endotracheal intubation and mechanical ventilation.1 Mechanical ventilation, particularly if prolonged, injures the lungs and contributes to BPD.2 Consequently, avoiding or minimising the time that extremely preterm infants are mechanically ventilated is critical.
The optimal way to provide respiratory support to extremely preterm infants after mechanical ventilation remains under investigation, and the transition from mechanical ventilation to non-invasive respiratory support remains a poorly understood process.2 There is a paucity of data on the optimal timing of extubation, criteria for readiness for extubation, and the best strategy to use when providing post-extubation respiratory support.3 The extubation failure rate in extremely preterm infants is high,4 and reducing this outcome must be a focus of research.
Nasal continuous positive airway pressure (CPAP) is the most frequently used mode of non-invasive support used after extubation of extremely preterm infants. The reasons for extubation failure during CPAP are multifactorial. Variables such as infant weight (birth weight <750 g), immaturity (<26 weeks’ gestation) and the severity of RDS (alveolar-arterial gradient >180 mm Hg) are weakly predictive of early CPAP failure in very preterm infants.5 The use of a set CPAP pressure sufficient to maintain functional residual capacity is likely to be important.6 The optimal CPAP pressure to use after extubation is unknown, although a meta-analysis of studies suggests that pressures of at least 5 cm H2O are needed.6 Many infants are reintubated following extubation for increased oxygen requirement and work of breathing suggesting that a low end-expiratory lung volume may contribute to extubation failure.5
Utilising higher CPAP pressures post-extubation may prevent alveolar collapse, improve lung function and reduce extubation failure.7 8 Kitsommart et al compared CPAP 7–9 cm water (H2O) with CPAP 4–6 cm H2O after extubation of infants with birth weight <1250 g and demonstrated no difference in extubation failure within 72 hours.7 In a second trial, Buzzella et al randomised very preterm infants born 23–30 weeks’ gestation with RDS to receive either CPAP 7–9 cm H2O or CPAP 4–6 cm H2O after extubation.8 Rates of extubation failure within 96 hours were significantly lower in the group randomised to the higher range of CPAP pressures.8 Current CPAP pressure recommendations are wide and varied.6 Most clinicians report pressures of 5–8 cm H2O, however, use of CPAP pressures up to 12 cm H2O have been reported and have not been associated with adverse effects.9
In extremely preterm infants, extubation failure is associated with significant morbidities, including BPD, pulmonary vascular disease, airway trauma, poor feeding and oral aversion, adverse neurodevelopmental outcomes and delayed family unit bonding.10 Thus, improving rates of successful extubation in this high-risk population of preterm infants is a clinical priority.10–14 The ÈCLAT trial will investigate the CPAP pressure range of 6–8 cm H2O, routinely used in our clinical practice, with a higher-pressure range of 9–11 cm H2O. We hypothesise that the higher-pressure range will result in less atelectatic pulmonary failure and extubation failure.
Methods and analysis
Study design and aim
We used the Standard Protocol Items: Recommendations for Interventional Trials checklist when writing our report.15 This is a multicentre, unblinded, randomised controlled trial. The aim of the ÉCLAT study is to determine, in extremely preterm infants born <28 weeks’ gestation who are undergoing their first extubation, whether extubation to a higher CPAP pressure (10 cm H2O, range 9–11 cm H2O), compared with a standard CPAP pressure (7 cm H2O, range 6–8 cm H2O) decreases extubation failure within 7 days.
Sample size
The rates of extubation failure within 7 days in extremely preterm infants at the participating centres is estimated at 55%. To detect a reduction in extubation failure from 55% to 35% (absolute risk reduction 20%, relative risk reduction 40%) with 80% power and a two-tailed alpha error of 0.05, a sample size of 93 infants in each arm (total 186 infants) is required.
Patient population
Infants born extremely preterm (<28 weeks’ gestation) who are intubated and mechanically ventilated and being extubated for the first time are eligible for participation in the ÉCLAT trial. The timing of the extubation is determined by the clinical team caring for the infant, and there is no postnatal age limit for participation.
Inclusion criteria
Infants are eligible if they
Were born <28 completed weeks’ gestation.
Are being extubated for the first time from mechanical ventilation to nasal CPAP.
Have received enteral or intravenous caffeine (as prophylaxis for apnoea of prematurity) <24 hours prior to the planned extubation.
Have received exogenous surfactant treatment.
Exclusion criteria
Infants are excluded if they
Are being extubated to any other mode of non-invasive respiratory support other than nasal CPAP, or to no respiratory support.
Have a major congenital anomaly or condition that might adversely affect breathing or ventilation: for example, known upper airway obstruction or major airway abnormality, or major congenital heart disease.
Are not receiving full intensive care after extubation.
Randomisation
Enrolled infants are randomised using Research Electronic Data Capture (REDCap) electronic data capture tools,16 hosted at the Murdoch Children’s Research Institute, Melbourne, Australia. REDCap is a secure, password-encrypted, web-based application designed to support data capture and randomisation for research studies. Only the infant’s first extubation is randomised. Multiple births are randomised individually. Randomisation occurs after the clinical decision to extubate has been made and shortly before extubation using a computer or smartphone. Stratification is by centre and gestational age at birth (<26 weeks; ≥26 weeks).
Clinical management
Higher CPAP pressure (intervention)
Infants are extubated to a set CPAP pressure of 10 cm H2O. While receiving CPAP, infants will remain within a set CPAP pressure range of 9 cm H2O–11 cm H2O for at least 24 hours, with changes within this range at the discretion of the treating team. After 24 hours, infants may have their set CPAP pressure weaned at the discretion of the treating team but must remain within a set CPAP pressure range 5 cm H2O–11 cm H2O for at least 7 days after extubation if receiving CPAP. Infants are reintubated if they satisfy the extubation failure criteria described below within 7 days after extubation. The fraction of inspired oxygen (FiO2) is titrated to keep oxygen saturations (SpO2) in the standard target ranges of the participating unit. If extubation failure occurs, management following reintubation will be at the discretion of the treating team. For subsequent extubations, clinicians will be encouraged to use the assigned set CPAP pressure range (see figure 1).

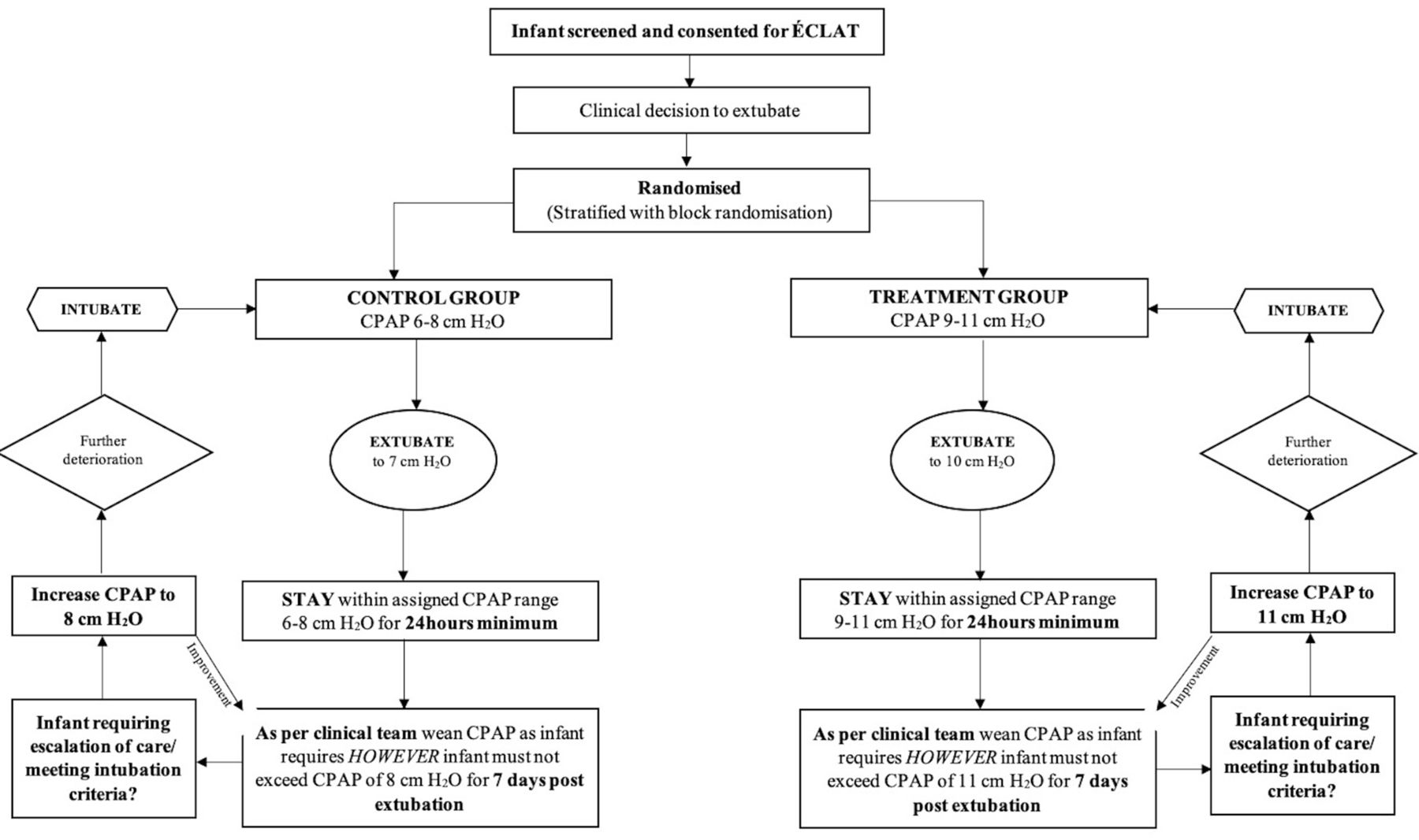{kind=link}
Study procedure. CPAP, continuous positive airway pressure; ÉCLAT, Extubation CPAP Level Assessment Trial.
Standard CPAP pressure (control)
Infants are extubated to a set CPAP pressure of 7 cm H2O. While receiving CPAP, infants will remain within a set CPAP pressure range of 6 cm H2O–8 cm H2O for at least 24 hours, with changes to the set CPAP pressure within this range at the discretion of the treating team. After 24 hours, infants may have their set CPAP pressure weaned at the discretion of the treating team but must remain within a set CPAP pressure range 5 cm H2O–8 cm H2O for at least 7 days after extubation if receiving CPAP. Infants are reintubated if they satisfy the extubation failure criteria described below within 7 days after extubation. The FiO2 is titrated to keep SpO2 in the standard target ranges of the participating unit. If extubation failure occurs, management following re-intubation will be at the discretion of the treating team. For subsequent extubations, clinicians will be encouraged to use the assigned set CPAP pressure range (see figure 1).
Device
In both groups, infants will be extubated to continuous-flow nasal CPAP, via a mechanical ventilator (either the Dräger VN500, Dräger Medical, Lübeck, Germany, or the SLE 5000, SLE, Croydon, UK) operating in CPAP mode. After 24 hours the infant may be transitioned to ‘bubble’ nasal CPAP (Fisher & Paykel bubble CPAP circuit, Fisher & Paykel Healthcare, Auckland, New Zealand) but only if receiving a CPAP≤10cmH2O given the pressure limitations of the ‘bubble’ CPAP device. Nasal CPAP may be delivered via any binasal CPAP prongs or mask, according to the participating unit’s protocol. Nasal prongs should be sized as per the manufactures guidelines to the largest size to occlude the infant’s nares.
Outcomes
Extubation failure
The primary outcome is extubation failure within 7 days, defined as receiving the maximum CPAP level (11 cm H2O in the intervention group; 8 cm H2O in the control group) and having at least one of:
FiO2 requirement >0.20 above the pre-extubation FiO2.
Two or more apnoeic episodes within any 24-hour period requiring intermittent positive pressure ventilation, or six or more apnoeic events requiring stimulation in any 6-hour period.
Respiratory acidosis with pH <7.2 and pCO2 >60 mm Hg.
Require urgent intubation for an acute deterioration (at clinical discretion) with the reason for reintubation documented.
Treatment failure
Should infants not be immediately reintubated and instead managed with non-invasive positive pressure ventilation or escalated to a higher CPAP pressure than their assigned range they will be documented as an extubation failure and reported as a protocol violation.
Secondary outcomes
Incidence of reintubation within 72 hours, and within 96 hours.
Failure in hours after extubation.
Reason(s) for extubation failure.
Kaplan-Meier Survival curve between both groups.
Death before hospital discharge.
Duration of mechanical ventilation in days after randomisation in survivors.
Total duration of hospitalisation in days in survivors.
Postmenstrual age at last supplemental oxygen, and at last positive pressure ventilation (mechanical ventilation, CPAP (or variants) or nasal high-flow >2 Litres per minute) in survivors.
Incidence of treatment with systemic postnatal corticosteroids for lung disease after randomisation.
Incidence of new pneumothorax requiring drainage with thoracocentesis or intercostal catheter insertion after randomisation.
Incidence of new, radiologically diagnosed pulmonary interstitial emphysema after randomisation.
Incidence of BPD, defined as a requirement for supplemental oxygen and/or respiratory support (mechanical ventilation, CPAP (or variants) or nasal high-flow >2 Litres per minute) at 36 weeks’ postmenstrual age.
Incidence of necrotising enterocolitis Bell’s stage 2 or above after randomisation.17
Incidence of spontaneous intestinal perforation after randomisation.
Incidence of retinopathy of prematurity requiring treatment with laser therapy or intraocular medication in one or both eyes after randomisation.
Incidence of new diagnosis of grade 3 or 4 intraventricular haemorrhage after randomisation.
Other data
Data collected will include
Maternal and infant demographics: maternal parity, infant sex, gestational age at birth, birth weight in grams, mode of delivery, exposure to any antenatal corticosteroids, duration of ruptured membranes prior to delivery in days, presence of histologically diagnosed chorioamnionitis.
Postnatal age at extubation in days, last weight prior to extubation in grams, age at first intubation in hours.
Previous dose of exogenous surfactant received in milligrams/kilogram, prior treatment for a patent ductus arteriosus (pharmacological or surgical), prior systemic postnatal corticosteroids for lung disease.
Mechanical ventilator settings immediately prior to extubation (mode, mean airway pressure in mm Hg, FiO2, tidal volumes (set and achieved), peak pressures (set and achieved) and end expiratory pressure.
Blood gas analysis results within 24 hours prior to extubation (if applicable): lowest pH, highest pCO2, lowest base excess.
Data analysis plan
Statistical analysis will follow standard methods for randomised trials. For the primary outcome, the analysis will be by intention to treat and be adjusted for the prerandomisation strata (gestational age (GA) and centre). For dichotomous outcomes, including the primary outcome, the two treatment groups will be compared using risk difference with 95% CI, both overall, and within the prespecified subgroups (gestational age at birth <26 weeks, ≥26 weeks). For dichotomous secondary outcomes, analysis will be limited to the two treatment groups, using risk difference with 95% CI. For continuous outcomes, the two treatment groups will be compared using difference of means, together with 95% CI, for outcome variables which are normally distributed; for outcome variables, which are not normally distributed, the comparison will be difference of medians, with 95% CI. All comparisons (risk difference, difference of means, difference of medians) will be estimated using regression models with the randomisation strata as covariates, and with SEs adjusted to take into account the clustering due to multiple births. Reporting of findings will be done in accordance with Consolidated Standards of Reporting Trials guidelines.
Adverse events
Adverse events (AEs) are recorded within 7 days after the randomised extubation. They are recorded as part of the study design and secondary outcomes of ÉCLAT. The site investigators are responsible for recording all AEs regardless of their relationship to the intervention. The following outcome are designated as AEs:
Necrotising enterocolitis (Bell’s stage III or IV).17
Intraventricular haemorrhage (grade III or IV).
Serious AEs
Serious AEs (SAEs) are recorded within 7 days after the randomised extubation. All are prespecified secondary outcomes of ÉCLAT. The investigators are responsible for recording all events regardless of their relationship to the intervention. All SAE’s are reported to an independent data safety monitoring committee (DSMC) and the local ethics committee within 72 hours of the principle investigator being notified. The following outcomes are designated as SAEs:
Death.
Spontaneous intestinal perforation.
Pneumothorax.
Pulmonary interstitial emphysema.
Study oversight
The independent DMSC established for the ÉCLAT trial have their roles and responsibilities detailed in a separate DSMC Charter. The DSMC includes two independent, experienced neonatologists and a senior statistician. The terms of reference for the DSMC include performance of interim safety analyses, periodic examination of relevant emerging external evidence, monitoring of AEs, compliance with the trial protocol, and progress of recruitment. Safety analyses by the DMSC are planned after the primary outcome is known for the first 50 and 100 infants and will occur blinded to group allocation. If required, an additional safety analysis will be performed at 150 infants. No interim analyses of the primary outcome are planned.
Clinical significance
Extubation failure is common in extremely preterm infants and associated with important neonatal morbidities.18 CPAP is the most commonly used form of non-invasive ventilation used postextubation but the optimal pressure to use for this indication remains uncertain. The ÉCLAT study will reveal novel information regarding CPAP pressures in extremely preterm infants. The ÉCLAT study is the largest trial comparing and researching CPAP pressures >8 cm H2O. Results from ÉCLAT will inform clinical practice and support clinicians in understanding and optimising CPAP pressures for extremely preterm infants. Results from this study will be disseminated via peer-reviewed journals and presented at national and international scientific conferences.
Supplemental material
Ethics statements
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @roseandtoby
Contributors RB, AM and AMK developed the concept and RB and AMK wrote the protocol. RB, BJM, RAB, AM and PGD gave input into the protocol and revised the manuscript. SMD designed the statistical analysis and revised the manuscript. All authors have read and approved the final manuscript and are accountable for its accuracy.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. However, AMK would like to thank the centre for research excellence in Newborn Medicine for their support NHMRC, GNT 1153176.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.