Article Text

Abstract
Introduction A novel method for the surgical treatment of benign prostatic hyperplasia (BPH) called Aquablation has become commercially available. Previous studies have been able to show similar functional results when compared with transurethral resection of the prostate and a high efficacy has been demonstrated when this approach is applied to patients with a prostate size of 80–150 cm3.
Holmium laser enucleation of the prostate (HoLEP) is a well-established procedure in the surgical treatment of BPH in prostate glands larger than 30 mL and a first-line therapy in glands over 80 mL. To date, no data are available whether Aquablation is non-inferior compared with HoLEP in the treatment of patients with medium-to-large-sized prostates regarding safety and efficacy.
Methods and analysis This is a prospective, randomised, open-label, non-inferiority clinical trial conducted at a Swiss centre of tertiary care. The primary outcome is assessment of non-inferiority of Aquablation compared with HoLEP in reducing lower urinary tract symptoms due to benign prostatic obstruction measured by the International Prostate Symptom Score (IPSS). Randomisation will be performed using secuTrial, stratifying on age (<70 years, 70+ years) and prostate volume (<100 mL, 100+ mL). Both interventions are performed in an inpatient setting and regular follow-up controls starting 8 weeks after intervention and continuing up to 5 years will be performed. The primary outcome (change in IPSS from baseline to 6 months) will be tested for non-inferiority with a one-sided t-test. Secondary outcomes, such as efficacy parameters, several patient-reported outcome measures, and periprocedural and safety parameters will be described by calculating means or relative frequencies for each treatment group and testing differences with two-sided standard superiority tests.
Ethics and dissemination The study was approved by the local ethics committee (EKOS 2020-02353). Results of the primary endpoint and each of the secondary endpoints will be published in an international peer-reviewed journal.
Trial registration number ClinicalTrials.gov Registry (NCT04560907).
- prostate disease
- surgery
- adult urology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the protocol for a prospective, randomised, open-label, non-inferiority clinical trial evaluating whether Aquablation is non-inferior compared with holmium laser enucleation of the prostate (HoLEP) regarding the reduction of patient-reported symptoms (lower urinary tract symptoms/benign prostatic obstruction (LUTS/BPO)) as measured by the International Prostate Symptom Score 6 months postoperatively.
Secondary outcomes include comparisons of several measures of relevance regarding surgical treatment of LUTS/BPO between Aquablation and HoLEP.
Regular follow-up controls starting 8 weeks after intervention and continuing up to 5 years will be performed for assessing both short-term and long-term treatment effects as well as complications.
The lack of blinding of the patients and assessors collecting postoperative data is a limitation of the study design.
Introduction
Benign prostatic hyperplasia (BPH) is one of the most common diseases in men and is often associated with bladder outlet obstruction and lower urinary tract symptoms (LUTS/BPO). Approximately 50% of men aged 50–60 years and ~90% of men aged ≥85 years are affected.1
Medical therapy is usually the first-line treatment.2 However, the efficacy of drugs like alpha-blockers is limited, and as disease progresses, more invasive treatment options have to be taken into consideration.
Holmium laser enucleation of the prostate (HoLEP) is an established procedure in the surgical treatment of BPH in prostate glands of 30 mL or larger and represents a first-line therapy in glands over 80 mL.3 The flat learning curve, operation time as well as the expensive equipment still limit the availability of HoLEP to a limited number of specialised centres and lead to the search for alternative surgical procedures.4 5
Recently, a new resective treatment technique of LUTS/BPO called Aquablation has become commercially available.6 This technique uses real-time ultrasound imaging combined with a robotically executed surgeon-guided high-velocity waterjet to resect prostate tissue. Previous studies have been able to show similar functional results when compared with transurethral resection of the prostate in a randomised study,7 and efficacy has also been demonstrated for patients with a prostate size of 80–150 cm3, claiming low operation time as well as a short hospital stay.8 Aquablation might offer its most relevant benefits in medium-to-large-sized prostates. However, no data comparing Aquablation with HoLEP in the treatment of patients with medium-to-large-sized prostates, which are regarded to be treated most appropriately by HoLEP nowadays, are available so far.
As Aquablation offers some obvious advantages compared with HoLEP regarding learning curve and operation time, the aim of this study is to test whether efficacy and safety of Aquablation are non-inferior compared with HoLEP in patients with medium-to-large-sized prostates, which would support the use of Aquablation in everyday clinical practice.
Objectives
Primary objective
To determine whether Aquablation is non-inferior compared with HoLEP regarding the reduction of patient-reported symptoms (LUTS/BPO) as measured by the International Prostate Symptom Score (IPSS) 6 months postoperatively.
Secondary objectives
Secondary objectives are to assess other factors that are important regarding BPH surgery and compare them between Aquablation and HoLEP. These factors include functional outcomes, patient-reported outcomes until 5 years after treatment, intervention and hospitalisation parameters, and treatment costs.
Safety objectives
Safety objectives of the study aim to compare both interventions regarding the nature, frequency and severity of treatment-related adverse events (AEs), and will be assessed applying the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) in grade 1–59 and a grading of complications according to Clavien classification.10
Methods and analysis
General study design and justification of design
This is a prospective, randomised, open-label, non-inferiority clinical trial conducted at the Department of Urology, School of Medicine, University of St Gallen, Switzerland. As mentioned before, the primary outcome is non-inferiority of Aquablation compared with HoLEP regarding reduction of LUTS/BPO.
The non-inferiority design was chosen as an efficacy of Aquablation similar of that of HoLEP would justify its use in clinical practice due to the shorter operation time and a faster learning curve.
At the same time, lack of detailed knowledge about the long-term outcomes of Aquablation and lack of comparative data of the two procedures regarding their relative merits and side effects justify the additional investigation of multiple secondary outcomes in a standard superiority setting.
Eligibility criteria
Inclusion and exclusion criteria are shown in table 1.
Inclusion and exclusion criteria
Randomisation
Dynamic treatment allocation will be performed through a data management program (secuTrial), which will be programmed and maintained by the Clinical Trials Unit (CTU) of Cantonal Hospital of St Gallen (KSSG). Randomisation will be performed using secuTrial, stratifying on age (<70 years, 70+ years) and prostate volume (<100 mL, 100+ mL).
A patient’s treatment will only be assigned after definite patient inclusion in the database and entry of baseline characteristics. This will guarantee treatment concealment during patient recruitment. Each patient will have an individual code/randomisation number, which has to be documented in the case report form (CRF). Randomisation will be initiated by authorised investigators of the trial after checking the informed consent and the inclusion and exclusion criteria.
Recruitment and screening
Recruitment of the study participants is performed in the outpatient clinic of the KSSG by authorised investigators who will check inclusion and exclusion criteria (table 1) by reviewing the patient’s medical record and by patient–doctor conversation.
Study participants are thoroughly informed about the study and possible questions are answered by the authorised investigators. If the patient feels well informed and confident to participate in the trial, informed consent can be given within the consultation.
Study interventions
After baseline visit, participants are randomised to HoLEP or Aquablation (figure 1). Both interventions are performed in an inpatient setting.

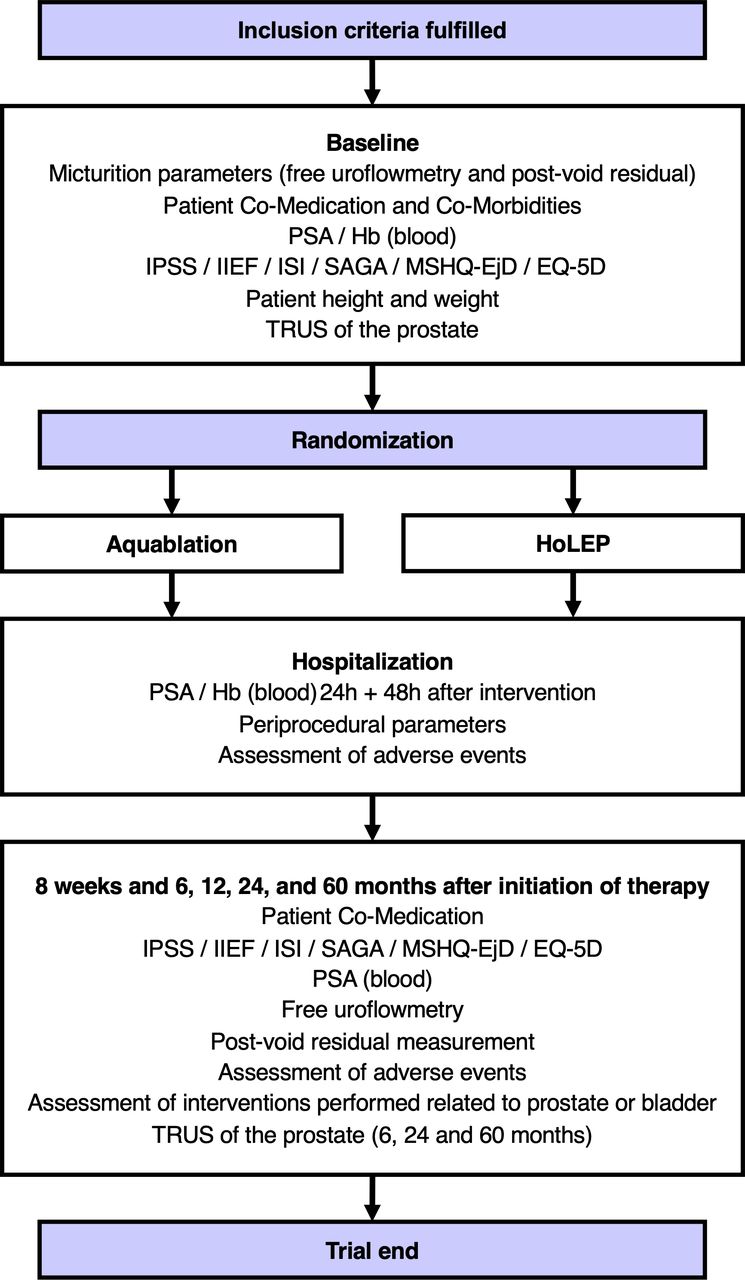{kind=link}
Flow chart. Hb, haemoglobin; HoLEP, holmium laser enucleation of the prostate; IIEF, International Index of Erectile Function; IPSS, International Prostate Symptom Score; ISI, Incontinence Severity Index; MSHQ-EjD, Male Sexual Health Questionnaire for Ejaculatory Dysfunction; PSA, prostate-specific antigen; SAGA, Self-Assessed Goal Achievement; TRUS, transrectal ultrasound.
Holmium laser enucleation of the prostate
The aim of HoLEP is to relieve pressure on the prostatic urethra by anatomically enucleating the majority of excess prostate tissue. This is done endoscopically under general or spinal anaesthesia. The three lobes of the prostate are enucleated en bloc with a holmium laser and then pushed into the bladder before being morcellated by a special instrument inserted through the resectoscope sheath. A catheter is placed through the urethra for bladder irrigation and left in place for around 24 hours before being removed on the day of discharge from hospital.
HoLEP is strongly recommended as a treatment option for moderate-to-severe LUTS with enlarged prostates by current guidelines.3 11
HoLEP represents a standard procedure at the Department of Urology of KSSG and will be carried out by experienced surgeons (minimum of 50 procedures)12 for the planned study.
Aquablation
Aquablation is a surgical technology for the therapy of benign prostate enlargement first introduced by Gilling et al6 in 2016 using the AquaBeam device (Procept BioRobotics, Redwood Shores, California, USA). The Aquabeam system includes a planning unit, a robotic handpiece and a surgeon console.13 By means of a high-pressure saline stream, parenchymal tissue of the prostate is removed endoscopically through a heat-free mechanism called hydrodissection under general or spinal anaesthesia. The intervention is supported by live ultrasound guidance, and the required depth as well as the angle of the resection is planned prior to the resection.13 The bladder is accessed using a 24-Fr handpiece, which accommodates the scope.14 The handpiece is supported by an articulating arm attached to the operation table. Once placed in the optimal position, the system automatically adjusts the alignment as necessary.13
Haemostasis is consecutively achieved through bipolar spot coagulation; a three-way catheter is inserted afterwards for bladder irrigation.
Aquablation was implemented in the clinical routine of the Urology Clinic in autumn 2019 and will be carried out by experienced surgeons (minimum of 20 aquablations)15 for the planned study.
If, during the course of the study, the company makes changes to the instruments or the procedure of the intervention, we will implement them according to the company’s recommendation.
Concomitant interventions
If an adaption, re-intervention or medication is indicated due to complications, side effects or patient’s preference, modifications, re-interventions and medication will be performed according to clinical practice and guideline recommendations.3
If symptoms that require a further treatment persist after Aquablation or HoLEP, medical, minimally invasive or surgical treatment options will be discussed with the patient. Performance and choice of treatment modifications depend on the type and degree of symptoms, inconvenience and patients’ preference.
In both groups, treatment changes and additional treatments will be assessed as a secondary study outcome.
Concomitant care and interventions are performed according to clinical practice and European Association of Urology guidelines recommendations.3
Characteristics and timing of visits
Regular follow-up controls starting 8 weeks after intervention and continuing up to 5 years will be performed as described in figure 1.
Study outcome measures
Primary outcome
Change in LUTS from baseline to 6 months after treatment measured by the IPSS.
Secondary outcomes
Efficacy parameters such as urinary flow and post-void residual urine and several patient-reported outcome measures assessed by validated and frequently used questionnaires, that is, prostate symptoms (IPSS), quality of life, sexual and erectile function (International Index of Erectile Function), Male Sexual Health Questionnaire for Ejaculatory Dysfunction, incontinence (Incontinence Severity Index) and Self-Assessed Goal Achievement (SAGA). Furthermore, laboratory parameters such as prostate-specific antigen, prostate volume measured by transrectal ultrasound, frequency of additional treatments (medical and surgical) and surgical re-interventions are assessed.
Periprocedural and safety parameters such as intervention characteristics (eg, procedural time, complications), hospitalisation parameters (eg, duration, post-procedure catheterisation, laboratory parameters) and AEs are also assessed.
In addition, other outcomes of interest such as the return to normal activity (EQ-5D questionnaire),16 treatment costs and cost-effectiveness as well as identification of potential predictors of therapy failure and success will be analysed.
Safety
All AEs are collected, fully investigated and documented in the source document and appropriate CRF during the entire study period, that is, from the patient’s informed consent until the last protocol-specific procedure, including a safety follow-up period.
In this study, AEs will be documented until 5 years after procedure. Assessment of AEs will be performed according to Clavien-Dindo classification10 and according to CTCAE V.59 as secondary study outcomes. Moreover, additional questions regarding complications and specific anamnesis within the scheduled control examinations are performed. Assessment of AEs will be performed during all scheduled and unscheduled visits. There will be no systematic assessment of AEs between scheduled controls.
Statistical methods
Hypothesis
Null hypothesis
The mean reduction in IPSS achieved with Aquablation until 6 months after start of treatment is smaller than the mean reduction achieved with HoLEP; the difference is at least equal to 4 points, which is assumed to represent a clinically relevant difference.17 18
Alternative hypothesis
The difference between the mean reduction in IPSS achieved with Aquablation and the mean reduction in IPSS achieved with HoLEP is smaller than 4 points (non-inferiority margin).
Sample size
Assuming a non-inferiority margin of 4 points, a true difference of 0 points, an SD of 6 points at each assessment and a correlation of 0.35 between assessments, 51 evaluable patients per group will provide 90% power to reject the null hypothesis in a t-test for non-inferiority at a one-sided significance level of 5%. To allow for 10% of dropouts, 114 patients need to be recruited. We aim to recruit 120 patients to allow for some uncertainty about the above assumptions.
Primary analysis
The primary outcome (change in IPSS from baseline to 6 months) will be tested for non-inferiority with a one-sided t-test assuming a non-inferiority margin of 4 points and a one-sided significance level of 5%. All patients who actually received the surgical treatment as randomised and who are evaluable for the primary outcome will be included (modified intention-to-treat principle).
This analysis will be performed after all included patients have performed the 6-month visit by the designated trial statistician.
Secondary analysis
Secondary outcomes will be described by calculating means or relative frequencies for each treatment group with 95% CIs. Differences between treatment groups will be tested with two-sided standard superiority tests that are appropriate for the distribution of each outcome (eg, t-tests, Wilcoxon rank-sum tests, χ2 tests, Fisher’s exact tests). By default, all patients who actually received the surgical treatment as randomised and with available data for a particular outcome will be included (modified intention-to-treat analysis). Additional analyses of alternative datasets will be performed if necessary.
Secondary analyses will be performed after all included patients have attended the 6-month visit, 2-year visit and 5-year visit (ie, separately at three times) by the designated trial statistician. All test results will be reported as numerical p values, and significance will be evaluated at a two-sided significance level of 5% without adjustment for multiple testing.
Missing data and dropouts
It is assumed that almost all patients will reach the primary endpoint. If patients drop out earlier, the dropouts should be offset by the planned number of 120 patients.
Ethics and dissemination
Ethics approval
The study was approved by the local ethics committee (EKOS 2020-02353). The study will be carried out in accordance to the protocol and with principles enunciated in the current version of the Declaration of Helsinki,19 the European Regulation on medical devices 2017/74520 and the ISO norm 14155 and ISO 14971,21 the Swiss Law and Swiss regulatory authority’s requirements.
Quality control, quality assurance and confidentiality
The Department of Urology of KSSG is instructed on standard operating procedures that are provided by the CTU on a regular basis.
Appropriate contracts with the CTU have been made and will guarantee quality assurances and control during the study, especially regarding data management, monitoring and randomisation.
Data collected specifically for the trial will not be included in the patient files but implemented directly into the study forms. Data from study forms will be transferred to an eCRF (secuTrial) on a regular basis.
All data, which are assessed independently from the study (eg, visit dates, demographical data, co-medication, interventional details, course of hospitalisation and so on) are stored in the medical history of the patient and transmitted into the CRF.
Data, which are assessed for study reasons only, are stored in the written study forms (eg, randomisation and unique patient number (UPN) form, AEs, serious AEs, concomitant medication) or in the trial master file (eg, informed consent, UPNs).
All data/documents concerning the study will be securely stored at our institution for 10 years, afterwards destroyed by shredding. The location of archiving will be the office of the sponsor-investigator at KSSG. Reports at the end of the study will be sent to the local ethics committee. The assessment and storage of all patient data will respect the Swiss data protection law and ISO 14155 norm.
Direct access to source documents will be permitted for purposes of monitoring, audits and inspections.
During and after the study, insight into the data collected in this trial will only be provided to the involved investigators and the members of the ethics committee if required.
Publication and dissemination policy
Results will be published in an international peer-reviewed journal and will be presented at urological congresses.
Participants will give consent for anonymised data sharing. Requests for an anonymised, full dataset of physician-level data and statistical code will be considered if the proposed use aligns with public good purposes, does not conflict with other requests, does not conflict with the planned use by the Trial Steering Committee, contingent on approval from the local ethics committee (EKOS). Requests can be addressed to the sponsor-investigator.
Patient and public involvement
The Department of Urology, School of Medicine, University of St Gallen, Switzerland, has an established and fruitful cooperation with patients interested in active trial participation: (https://www.kssg.ch/urologie/lehre-forschung/patienten-und-oeffentlichkeitsbeteiligung-der-planung-klinischer-studien).
Out of this cooperation, we were able to learn that standardised patient-reported outcome measures (ie, questionnaires) and voiding parameters often do not address the main concern of individual patients appropriately. Therefore, we included the SAGA22 in the present trial. Thus, patients can define their main treatment goals prior to treatment, and achievement of goals according to a Likert scale is assessed at each study visit.
Discussion
The aim of this study is to assess whether Aquablation is a valuable treatment option compared with HoLEP in patients with LUTS/BPO and medium-to-large-sized prostates, assessing both short-term and long-term treatment effects as well as complications. Using a prospective, randomised, non-inferiority trial design with clearly defined outcomes, as well as inclusion and exclusion criteria, and performed according to well-defined quality standards, data will help to better estimate treatment efficacy and safety of Aquablation. Furthermore, potential benefits as well as problems could be analysed to show if Aquablation is potentially an equivalent alternative to HoLEP in the treatment of patients with medium-to-large-sized prostates, which would support the use of Aquablation in daily clinical practice.
References
Footnotes
Contributors GM, DA and SG participated in creating the study design. GM and DA drafted the manuscript. H-PS, VZ, PB, DSE and SG provided a critical revision of the manuscript. GM and DA obtained the funding of this study. All the authors read and approved the final manuscript.
Funding The trial will be supported by a grant from the Blumenau-Léonie-Hartmann Foundation.
Disclaimer The funder will have no role in the conduct or analysis of the trial.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.