Article Text

Abstract
Introduction Tinnitus is the perception of sound without an external stimulus, often experienced as a ringing or buzzing sound. Subjective tinnitus is assumed to origin from changes in neural activity caused by reduced or lack of auditory input, for instance due to hearing loss. Since auditory deprivation is thought to be one of the causes of tinnitus, increasing the auditory input by cochlear implantation might be a possible treatment. In studies assessing cochlear implantation for patients with hearing loss, tinnitus relief was seen as a secondary outcome. Therefore, we will assess the effect of cochlear implantation in patients with primarily tinnitus complaints.
Method and analysis In this randomised controlled trial starting in January 2021 at the ENT department of the UMC Utrecht (the Netherlands), patients with a primary complaint of tinnitus will be included. Fifty patients (Tinnitus Functional Index (TFI) >32, Beck’s Depression Index <19, pure tone average at 0.5, 1, 2 kHz: bilateral threshold between ≥40 and ≤80 dB and hearing thresholds in the ear to be implanted (≥4 kHz) ≥50 dB) will be randomised towards cochlear implantation or no intervention. Primary outcome of the study is tinnitus burden as measured by the TFI. Outcomes of interest are tinnitus severity, hearing performances (tinnitus pitch and loudness, speech perception), quality of life, depression and patient-related changes. Outcomes will be evaluated prior to implantation and at 3 and 6 months after the surgery. The control group will receive questionnaires at 3 and 6 months after randomisation. We expect a significant difference between the cochlear implant recipients and the control group for tinnitus burden.
Ethics and dissemination This research protocol was approved by the Institutional Review Board of the University Medical Center (UMC) Utrecht (NL70319.041.19, V5.0, January 2021). The trial results will be made accessible to the public in a peer-review journal.
Trial registration number Trial registration number NL8693; Pre-results.
- otolaryngology
- audiology
- adult otolaryngology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The randomised controlled study allows for high quality assessment of outcomes of cochlear implantation for patients suffering primarily from tinnitus and secondarily from moderate to moderately severe bilateral hearing loss.
Outcomes of interest are not limited to tinnitus burden but also consider anxiety and depression, quality of life and patient-related changes.
The intervention can induce risks associated with surgery and a residual hearing deterioration in the ear implanted which will be monitored by electrocochleography measurement.
This study is a further step towards evidence-based medicine for the clinical efficacy of cochlear implants as a tinnitus treatment.
Background
Tinnitus is the perception of sound without an external stimulus, often experienced as a ringing or buzzing sound.1 2 It is a common symptom with an approximate prevalence of 10%–30%, depending on the selected population,3 increasing to 30% of adults over the age of 50 years.4 Tinnitus can be chronic and disabling for those individuals affected by it. It is a complex condition, in which many components are responsible for perceived burden, like loudness, comorbidity and sleep problems. The heterogeneous aspect of the disease is also accountable for differences in the tinnitus itself: localisation, sound characteristics, temporal course and underlying cause. The tinnitus burden and the individual needs of patients for tinnitus-related healthcare are various. While the underlying aetiology of tinnitus is still debated, one hypothesis is that the tinnitus arises from changes in neural activity caused by reduced or lack of auditory input due to hearing loss which often accompanies the tinnitus.5 6 To date, the only evidence-based therapy for the reduction of tinnitus burden is cognitive behavioural therapy5 7–9 which is offered as standard clinical care in many countries in people with bothersome tinnitus.10 However, this therapy only improves tinnitus distress but does not reduce tinnitus loudness.11 Sound therapy is also considered as a recommendation for patients with hearing loss according to European guidelines but there is a lack of conclusive evidence.10 12 13
Since auditory deprivation is thought to be one of the causes of tinnitus, increasing the auditory input by cochlear implantation might be a possible treatment option. This hypothesis is confirmed by observations in studies assessing the effectiveness of cochlear implantation to restore hearing function in case of bilateral deafness, where tinnitus reduction is one of the secondary outcomes.14 Analysing the effect of intracochlear electrical stimulation with a cochlear implant (CI) on primarily tinnitus complaints has been investigated by only few studies. All studies assessing the effect of cochlear implantation for tinnitus concerned cases with single-sided deafness15–20 or patients with asymmetrical hearing loss.6 They all reported a significant tinnitus reduction after implantation. So far, there is no high level of evidence of the effect of intracochlear stimulation as an intervention for primary tinnitus complaint in case of bilateral moderate to severe hearing loss.14
Above mentioned studies provide the first evidence of possible effectiveness of cochlear implantation for the reduction of tinnitus burden. To provide clear evidence of the effectiveness of cochlear implantation for the suppression of tinnitus complaints, a statistically powered study is needed aiming at patients with tinnitus as their primary complaint instead of hearing loss. To what extent electrical stimulation can reduce tinnitus in patients with bilateral moderate to severe hearing loss (just below the current CI indication), but with primary complaint of tinnitus, is unknown.21 Therefore, we aim to study the effect of cochlear implantation on tinnitus burden in patients suffering primarily from tinnitus and failed standard clinical care. For these patients which also have a bilateral moderate to severe hearing loss, a randomised controlled trial (RCT) will be conducted in which cochlear implantation will be compared with no intervention.
Method and analysis
Study objectives
The primary objective of this study is to assess the effect of electrical stimulation by a CI on tinnitus burden, measured with the Tinnitus Functional Index (TFI) at 6 months after cochlear implantation. Secondary outcomes are to assess the effect of CI on tinnitus severity, tinnitus pitch and loudness, auditory function, speech recognition, quality of life, symptoms of depression and anxiety, patient reported change in order to attest treatment-related differences.
Patient involvement
Patients were not involved in the design, or conduct, or reporting or dissemination plans of the study.
Study design and setting
The study is a monocenter clinical trial performed in a tertiary referral clinic (university hospital) in the Netherlands (University Medical Center Utrecht). The protocol is reported according to the Standard Protocol Items: Recommendations for Interventional Trials statement.22 In this RCT, patients will be randomised into groups: a CI group and a control group (figure 1). Twenty-five patients (CI group) shall receive a CI in the ear mostly affected by tinnitus. The other 25 patients (control group) shall follow a follow-up period of 6 months with no intervention. The follow-up sessions will take place 3 and 6 months after implantation to assess the primary outcome of tinnitus burden and secondary outcomes of quality of life, treatment-related outcomes and auditory function.

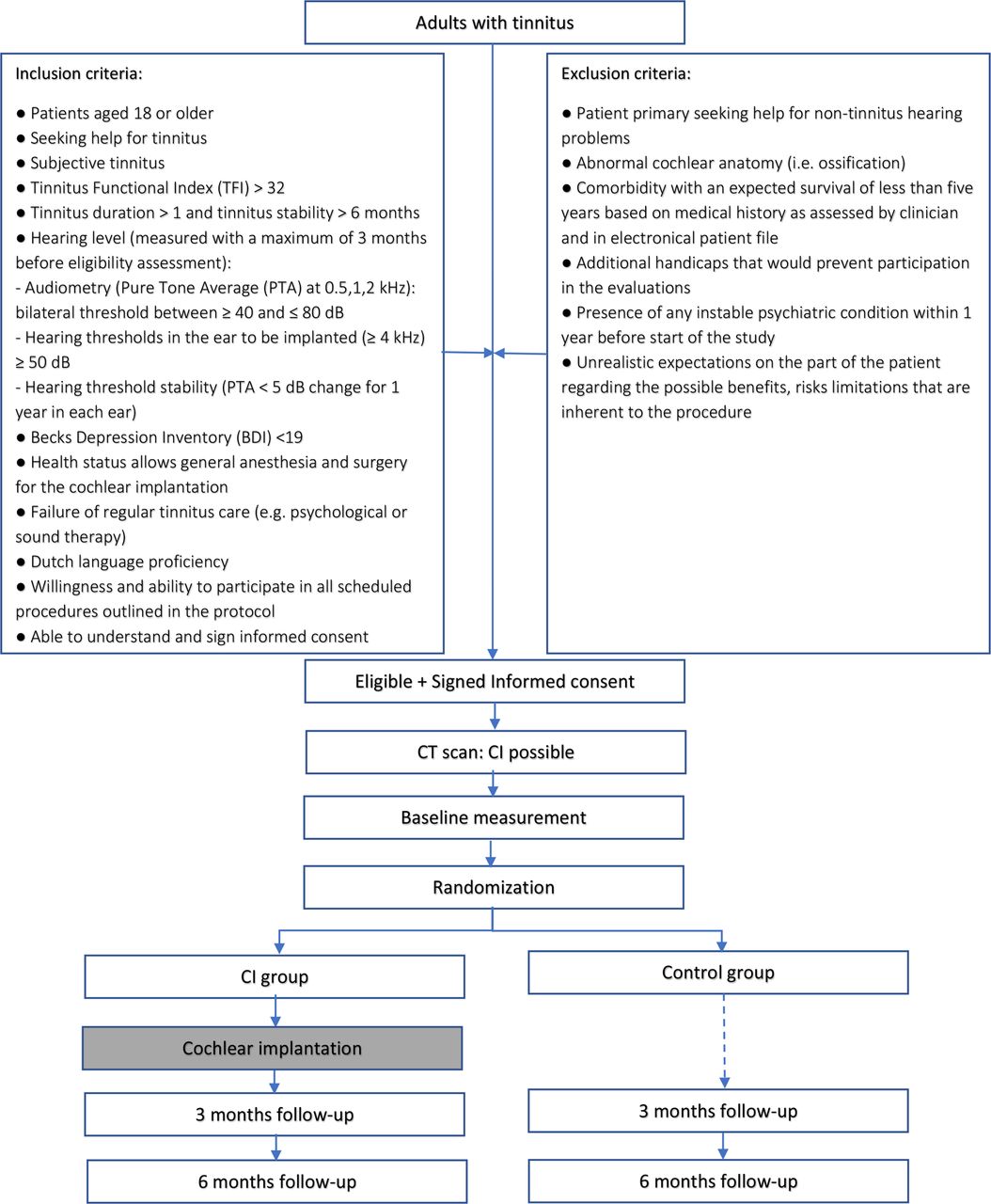{kind=link}
Study flowchart. CI, cochlear implant group; control, control group.
Study population
The study population consists of patients seeking help for tinnitus, presenting at the outpatient clinic of Ear, Nose and Throat (ENT) of the UMC Utrecht, The Netherlands. Fifty patients aged 18 or older with moderate to severe tinnitus and moderate to severe hearing loss will be included after fulfilling eligibility and informed consent. They must meet the following criteria to be eligible for the study at randomisation.
Inclusion criteria
The eligibility criteria for patients are:
Patients aged 18 or older.
Seeking help for tinnitus.
Subjective tinnitus.
Moderate to catastrophic tinnitus burden: TFI >32.
Tinnitus duration >1 year and tinnitus stability >6 months.
Hearing level (measured with a maximum of 3 months before eligibility assessment):
Audiometry (pure tone average (PTA) at 0.5, 1, 2 kHz): bilateral threshold between ≥40 and ≤80 dB.
Hearing thresholds in the ear to be implanted (≥4 kHz) ≥50 dB.
Hearing threshold stability (PTA <5 dB change for 1 year in each ear).
No to mild depression: Becks Depression Inventory (BDI) <19.
Health status allows general anaesthesia and surgery for the cochlear implantation.
Failure of regular tinnitus care (eg, psychological or sound therapy).
Dutch language proficiency.
Willingness and ability to participate in all scheduled procedures outlined in the protocol.
Able to understand and sign informed consent.
Exclusion criteria
A potential patient who meets any of the following criteria will be excluded from participation in this study:
Patient primary seeking help for non-tinnitus hearing problems.
Abnormal cochlear anatomy (ie, ossification).
Comorbidity with an expected survival of less than 5 years based on medical history as assessed by clinician and in electronical patient file.
Additional handicaps that would prevent participation in the evaluations.
Presence of any unstable psychiatric condition within 1 year before start of the study.
Unrealistic expectations on the part of the patient regarding the possible benefits, risks and limitations that are inherent to the procedure.
If a patient is eligible for the study, his/her otorhinolaryngologist will ask him/her to participate. The content of the study will be explained by the patient’s otorhinolaryngologist who will provide him/her written patient information and the informed consent form. Patients will be given 2 weeks to consider participation. If a patient meets the criteria for inclusion and exclusion and wants to take part in the study, the patient will be asked to come to the UMC Utrecht for a CT scan to visualise the anatomy of the mastoid. If the patient’s CT scan shows normal cochlear anatomy, he will, during the same visit, sign the informed consent with a member of the research team and receive a copy of the consent. After inclusion, baseline measurement will be performed where after randomisation will take place.
Recruitment status and trial dates
Patient enrolment started in January 2021 and will be completed in June 2022. The surveys and measurement will be performed until January 2023.
Randomisation
After inclusion and baseline measurement, patients will be randomly allocated into one of the two groups: CI group or control group. The randomisation will be computer-generated with block sizes of 4 and 6 and stratified for TFI score. A website randomisation programme, developed by Castor EDC23 will be used for randomisation. A study database was set up in Castor EDC to support allocation and concealment. Investigators enter information for each eligible patient and the randomisation assignment is revealed once the investigators validate the inclusion of the patient. The block design is unavailable to those who assign participants until the moment of assignment. Blinding is not possible during this study since both patients and caregivers will be able to see from outside whether patients have a CI or not.
Intervention
Patients allocated to the intervention group will receive a CI. The CI will be implanted on the most affected tinnitus side, and if equal tinnitus in the two ears, in the ear with the worst hearing loss. Hearing aid will be allowed in the contralateral ear. The cochlear implantation will be carried out under general anaesthesia after consent of the anesthesiologist and after determination of general health status. The standard surgical procedures for cochlear implantation will be followed. A retro-auricular incision will be made to expose the mastoid. The electrode will be inserted via a posterior tympanotomy and round window implantation by soft-surgery techniques. Intraoperatively, normal functioning of the device will be checked by measurement of impedance and neural response telemetry. Electrocochleography will also be recorded intraoperatively using Cochlear Research Platform (V.1.1). The CI used for the study consists of a Nucleus 7 sound processor and a CI622 implant with a slim straight electrode from Cochlear (or similar). Serial numbers of the CIs will be registered in the operating room report by the surgeon (standard clinical care for cochlear implantation) and in the master study file (product accountability). A postoperative cone beam CT of the mastoid will be planned to detail the electrode location within the cochlea.
One week after surgery patients from the intervention group will be checked at the outpatient department of the ENT to check for wound healing. The rehabilitation phase will start 4 weeks after surgery with a visit of the patient to the department of audiology to custom fit the processor software and then (bi)weekly till week 11 after surgery to fine tune the programming of the implant and improve speech perception. The CI fitting will not differ from the standard of care and will be optimised for every patient.
In the follow-up phase, the patients with CI will return to the UMC Utrecht 3 and 6 months after implantation to assess study outcome by the research team. The patients of the control group will come to the UMC Utrecht 3 and 6 months after randomisation to assess the same study outcome. A questionnaire will have to be filled in at home by the patients before every follow-up session at 3 and 6 months, as well as 2 weeks after surgery for the intervention group.
Participants are not allowed to start another tinnitus treatment during the study.
Sample size
To detect a clinically relevant difference of one grade (15 points) change measured with the TFI,24 in tinnitus burden at 6 months after cochlear implantation compared with the control group, with a power of 90% and alpha of 0.05, 23 patients are needed in both arms of the study. An acceptable SD was set at 15, based on the results of a previous pilot study assessing CI for tinnitus patients.20 We will include 25 patients per arm, a 10% margin, to include for possible lost to follow-up. Thereby, we expect patients to have a mean TFI at baseline of 50 points on TFI (grade 3) and a TFI decrease of 15 points at 6 months after intervention with a mean endpoint of 35 points on TFI (grade 2).
Outcomes
The following outcomes will be assessed at the baseline visit and follow-up visits at 3 and 6 months after randomisation (table 1). All measurements will be performed by the research team following the same protocol procedures.
Schedule of visits and assessments to measure study outcome per group
Primary outcome measure
Our primary outcome is tinnitus burden as measured with the validated TFI. The TFI is a 25-item containing questionnaire with statements/questions about tinnitus burden.24 25 The index is divided in eight subscale items: intrusive, sense of control, cognitive, sleep, auditory, relaxation and quality of life. Possible answers are ranging between 0 and 10, resulting in a maximum score of 100, representing a maximum burden of tinnitus. This total score is then categorised into five different grades, indicating low to high burden.
Secondary outcome measures
Audiological tests
Five audiological measurements are included in the study and are performed by an audiologist according to the ISO 16832:2006.26
Pure tone audiometry
The first evaluation is a pure tone audiometry at 0.25, 0.5, 1, 1.5, 2, 4 kHz. This standard measurement evaluates the audible threshold of the patient by having patients indicating audibility for frequency-specific pure tone stimuli at different loudness level. The evaluation results in an audiogram which provides information about the hearing level of the patients.
Speech recognition test in quiet and noise
The second evaluation is a speech recognition test in quiet and noise. For the patients receiving a CI, post-intervention assessments will be applied with the CI. The participant is listening at digits, Dutch words and sentences in a sound-treated booth. The loudness of the speech will change during the test in steps of 2 dBs, but the noise signal will be presented at a constant level of 65 dB Sound Pressure Level (SPL). The patient is asked to repeat back the words. The patient will perform the same test in two different conditions: with or without noise. This test results in a speech reception threshold obtained by averaging the signal-to-noise ratio over the list of words presented in order to obtain a 50% correct score. The outcome will permit to set up a rehabilitation programme with a speech therapist for the intervention group.
Electrocochleography
Electrocochleography (ECochG) is a technique to record electrical potentials generated in the inner ear and auditory nerve in response to acoustic stimulation. ECochG measurement will be performed intraoperatively and at 3 and 6 months after cochlear implantation. The measure will be followed by conventional audiological examination. During the measurement postoperatively, the patient will be asked to sit comfortably on a chair and not move. The operator will install the earplug in the patient’s ear and connect it to an audio cable attached to a sound processor. The sound processor will generate acoustic stimulation through the audio cable and the electrical responses will be recorded in real time via the Cochlear Research Platform (V.1.1, Cochlear ltd). The ECochG provides a measure of the cochlear function.
Pitch match experiment
Pitch match of tinnitus is performed to find the pitch corresponding to the tinnitus pitch of the patient. An acoustic pitch matching and an electric pitch matching will be performed in a sound-treated booth. The acoustic pitch matching will provide information about the frequency of the tinnitus perceived whereas the electric pitch matching will provide information about the pitch-matched electrode. The patient will be asked to concentrate on the predominant pitch of their tinnitus. Two tones will be presented at the same intensity level previously matched with tinnitus. The patient will indicate which option, the first or the second, sounds the closest in pitch by manipulating the response switch forward and backward. The difference between the first and the second will become smaller and smaller, until there is one frequency that matches best. Each stimulation will be performed two times (apical-to-basal and basal-to-apical to prevent octave-confusion). The pitch matched will be identified as the pitch resulting of the two runs. If the result of the two runs is not the same, the procedure will be repeated until finding a consistent result at least two times.27
Loudness match experiment
Loudness match of tinnitus is performed to find the loudness corresponding to the tinnitus acoustically and electrically.28 The experiment uses the tinnitus pitch matched. The pure tones are initially presented at 6 dB above threshold. The patient is instructed to adjust the loudness of the comparison tones to match that of their tinnitus. The adjustment of the intensity is made in a range of 5 dB for rough determination and then 1 dB steps until a satisfactory loudness match in obtained.
CI usage
The history of several user characteristics will be logged from the processor. This provides the following outcome parameters:
Time on air, providing the time the device was used in speech environment or the device was off.
Scenes, providing the time spending in different environments: quiet, speech, noise, speech in noise, music and wind.
Level of the environmental sound in dBA.
Programme usage, providing a daily average on programme usage.
Questionnaires
Questionnaires will be sent by email to the study participants through the data management programme Castor EDC.23 If participants do not want to perform online questionnaires, they will receive paper versions of the questionnaires by postal services. All questionnaires will be in the Dutch language.
Tinnitus questionnaire
The Visual Analogue Scale (VAS) tinnitus has two items. The patient answers two questions about tinnitus severity and intrusiveness using a visual analogue scale that ranges from 0 (not at all) to 10 (extremely).
Tinnitus history
The ESIT Screening Questionnaire (ESIT-SQ)29 consists of 39 items relevant for tinnitus profiling including 17 general and 22 tinnitus-specific questions. Every question presents multiple choice. The test is used a baseline questionnaire and takes approximately 10 min to fill in.
Patient reported benefits
The Clinical Global Impression (CGI) consists of a one-item observer-rated scale that measures global improvement or change (CGIC).30 The question is scored on a scale from 1 to 7, 1 meaning ‘very much improved’ to 7 meaning ‘very much worse’.
The Glasgow Benefit Inventory (GBI) is a validated questionnaire reporting change in health-related quality of life postintervention.31 It consists of 18 questions scored on a 5-point Likert scale where 1 indicates ‘much worse’ and 5 is for ‘much better’. The questionnaire presents three different items: general subscale, social support and physical health.
Quality-of-life questionnaires
The Euro-Quality-of-life 5D (EQ5D) is a standardised measure of generic health status. It contains only five questions. Each question deals with a specific domain: mobility, self-care, usual activities, pain/discomfort and anxiety/depression.32 The patient must choose between different sentences which corresponds to his/her health condition. The last question is a self-report of the overall health status using a visual analogue scaling from 0 (the worst health you can imagine) to 100 (the best health you can imagine).
The Speech, Spatial and Qualities Hearing Scale (SSQ) measures hearing-related quality of life and consists of three scales that assess different domains of hearing: (1) the speech hearing subscale consists of 15 questions that assess the ability to separate speech from competing noise in a wide range of listening contexts, (2) the spatial hearing subscale consists of 17 questions that assess the ability to locate sound sources and their direction of movement, (3) the quality of hearing subscale consists of 19 questions that assess naturalness and clarity of sound sources.33 Possible answers are scored using a visual analogue scale ranging from 0 (not at all) to 10 (excellent).
Comorbid symptom scores
The BDI is a 21-item questionnaire used as an indicator of the severity of depression.34 Each question is scored on four points ranged between 0 (for example ‘I do not feel sad’) and 3 (‘I am so sad’) with a maximum of total score of 63.
The Hospital Anxiety and Depression Scale (HADS) is a 14-item screening tool for anxiety and depression symptoms in non-psychiatric clinical populations.35 36 Each sentence is scored between 0 and 3 where 0 confirms the sentence and 3 disagrees with it.
Statistical analysis
Baseline characteristics per group will be described as means or medians, depending on the normality of the data and SD. Between-group mean differences will be calculated with 95% CIs. A p value <0.05 is considered statistically significant.
The primary outcome will be the difference in TFI score between the intervention at 6 months after cochlear implantation and the control group after 6 months of no intervention, a continuous variable. Differences between the control and intervention group will be calculated using the unpaired t-test and the Mann-Whitney U test. The secondary outcome measures will be the performances on the auditory tests and the questionnaires. Differences between groups will be calculated using the unpaired t-test and the Mann-Whitney U test. Within-subject comparisons will entail differences of mean values. These will be analysed using paired t-tests for continuous measures.
Interim analyses on the safety data will be performed and reviewed by an external data safety monitoring board (DSMB). An interim analysis will be done every 6 months starting after the five first patients reached 6 months of follow-up. A statistician will perform non-parametric test on the aided speech perception of the implanted ear only, performed 6 months postimplantation to monitor functional hearing performance. The DSMB will advise on stopping the study if there is a risk for the patient’s safety based on tinnitus worsening and deterioration of functional hearing.
Potential missing data will be handled using multiple imputation. Complete cases analyses will be done as a sensitivity analysis. All analyses will be performed on an intention-to-treat basis.
Ethics and dissemination
Protocol version
The study will be conducted according to the principles of the Declaration of Helsinki (version 2013, Fortaleza) and in accordance with the Medical Research Involving Human Subjects Act (WMO). The research protocol was approved by the Institutional Review Board (IRB) of the UMC Utrecht (NL70319.041.19) and the Dutch competent authorities.
Protocol amendment
All amendments will be notified to the local Medical Research Ethics Committee (MREC). The data from this study will be used for publication in peer-reviewed international journals, preferably open-access. To diminish possible chance on publication bias, the study will be reported using the Consolidated Standards of Reporting Trials guidelines.37
Confidentiality
All data will be treated confidentially. The data will be encrypted by using an unique patient identification number. The analysis will be performed with these coded patient data. The key code will be safeguarded by the investigators. The paper data files and informed consents will be stored in a locked cabin in a locked room. The data will be stored on the investigator’s computer as well, which is secured by a password and situated in a locked room. The handling of personal data will comply with the EU General Data Protection Regulation and the Dutch Act on Implementation of the General Data Protection Regulation, the Uitvoeringswet AVG, UAVG. The final trial dataset will be safeguarded and available to the principal investigator and approved members of the research team.
Data monitoring and auditing
The investigator will submit a summary of the progress of the trial to the accredited MREC once a year. Information will be provided on the date of inclusion of the first subject, numbers of subjects included and numbers of subjects that have completed the trial, serious adverse events (SAEs)/serious adverse reactions, other problems and amendments. Trial quality will be monitored independently by the Julius Clinical Centre (UMC Utrecht, the Netherlands) according to regulations by the UMC Utrecht and the Dutch government. The local monitor will check 50% of signed informed consents, inclusion and exclusion criteria, source data and SAEs. Due to the high-risk nature of the study, an external DSMB will be in place to perform ongoing safety surveillance. An interim analysis will be performed by the statistician of the research group and will be analysed by the DSMB every 6 months after the fifth first inclusions.
Adverse events
Besides the normal risks associated with surgery and general anaesthesia, adverse events related to cochlear implantation will be monitored by assessment and documentation of intraoperative and postoperative complications and device failures. Deterioration of the hearing <30 dBs (PTA) is expected after implantation because of the cochlear trauma and should not be considered as an adverse event.38 39 All adverse events will be followed until they have abated or until a stable situation has been reached. All cases of SAEs will be reported to the local IRB and the Dutch competent authorities.
Trial status
The study is currently in recruitment phase.
Ethics statements
Patient consent for publication
Ethics approval
This research protocol was approved by the Institutional Review Board (IRB) of the UMC Utrecht (NL70319.041.19, V5, January 2021).
References
Footnotes
Contributors All authors (KA, ALS, IS, KSR, RS and BvD) developed the protocol. IS provided statistical expertise in clinical trial design. KA drafted the manuscript. All other authors revised themanuscript. All authors read and approved the final version.
Funding Part of cost involved of this study is funded by Cochlear Ltd. as a non-restrictive research grant (IIR1975). Cochlear Ltd. did not and will—by a research contract—not have influence on the data collection, analysis, data interpretation and publication.
Competing interests KA received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant (agreement number 764604). KA and BvD are employed at Cochlear Technology Centre, Mechelen, Belgium.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.