Article Text

Abstract
Introduction Immune-mediated inflammatory diseases (IMIDs) are associated with reduced health-related quality of life (HRQol), increased risk of somatic and psychiatric comorbidities and reduced socioeconomic status. Individuals with one IMID have an increased risk for developing other IMIDs. The unmet needs in the care of patients with IMIDs may result from a lack of patient-centricity in the usual monodisciplinary siloed approach to these diseases. The advantages of novel interdisciplinary clinics towards the traditional therapeutic approach have not been investigated. The overall aim of this study is to determine the effectiveness of an interdisciplinary combined clinic intervention compared with usual care in a population of patients with the IMIDs: psoriasis, hidradenitis suppurativa, psoriatic arthritis, axial spondyloarthritis and inflammatory bowel disease. Our hypothesis is that an interdisciplinary combined clinic intervention will be more effective than usual care in improving clinical and patient-reported outcomes, and that a more effective screening and management of other IMIDs and comorbidities can be performed.
Methods and analysis This is a randomised, usual care controlled, parallel-group pragmatic clinical trial. 300 consecutively enrolled participants with co-occurrence of at least two IMIDs are randomly assigned in a 2:1 ratio to either treatment in the interdisciplinary combined clinic or usual care. The study will consist of a 6-month active intervention period and a 6-month follow-up period where no intervention or incentives will be provided by the trial. The primary outcome is the change from baseline to 24 weeks on the Short-Form Health Survey (SF-36) Physical Component Summary. Additional patient-reported outcome measures and clinical measures are assessed as secondary outcomes.
Ethics and dissemination Ethical approval of this study protocol was established by the institutional review board of the study site. The findings from this trial will be disseminated via conference presentations and publications in peer-reviewed journals, and by engagement with patient organisations.
Trial registration number NCT04200690.
- inflammatory bowel disease
- psoriasis
- rheumatology
- immunology
- therapeutics
- organisation of health services
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- inflammatory bowel disease
- psoriasis
- rheumatology
- immunology
- therapeutics
- organisation of health services
Strength and limitations of this study
This is the first randomised, usual-care controlled trial to assess the effectiveness of a coordinated interdisciplinary approach to disease management in patients with immune-mediated inflammatory diseases (IMIDs).
The focus of the study will be on personalised, preventive and participatory healthcare.
The pragmatic elements in the design of this trial increase the likelihood that the results can be generalised to everyday practice and support decision-making by patients, providers and health system leaders.
Emphasis on generic patient-reported outcome measures that can be used across age, disease and treatment groups enables a meaningful assessment of patients with complex IMIDs and creates a strong focus on patient-centricity.
Investigators and patients cannot be blinded to participation randomisation outcomes due to pragmatic design limitations.
Introduction
Immune-mediated inflammatory diseases (IMIDs) including autoimmune diseases affect up to 10% of the western population.1 Among these are inflammatory bowel diseases (IBD) including ulcerative colitis (UC) and Crohn’s disease (CD), spondyloarthritis (SpA) including axial spondyloarthrtis (axSpA), ankylosing spondylitis (AS) and psoriatic arthritis (PsA), psoriasis and hidradenitis suppurativa (HS). The aetiology of IMIDs is only scarcely understood, but known to consist of a combination of genetic susceptibility and dysfunctional immunological mechanism resulting in a loss of immunological tolerance towards specific tissues, with a considerable overlap in organ involvement between the different disease types. The diseases listed above are all associated with cardiometabolic disease, malignancy, infections, ophthalmologic diseases, psychiatric disorders and reduced socioeconomic status.2–6 An association between several of the diseases has been shown.7–11 Additionally, it is generally accepted that individuals with one IMID have an increased risk for developing other IMIDs.
Despite this knowledge, a number of challenges currently exist in providing high-quality care for patients with co-occurrence of more than one IMID. These challenges include limited awareness of other autoimmune diseases among patients and healthcare professionals (HCPs); lack of screening for other autoimmune diseases; unidisciplinary siloed approach to care; delayed referral from one specialist to the next one, lack of consensus regarding treatment goals and outcome measures; lack of patient-centricity; unrecognised, underdiagnosed and undertreated comorbidities; and lack of regular follow-up.12
The above-mentioned siloed approach to care may lead to a lack of patient-centricity and inefficient management of the disease. In a Danish qualitative study, it was reported that some patients experience lack of physician continuity, lack of communication between various HCPs, a need for patients to relay health-related information between various HCPs, contradicting information about disease activity from various HCPs, work-related uncertainties, a lack of knowledge and disease understanding in the social system and negative consequences in the social system of the delayed diagnostic process.13 14
Recent retrospective studies have reported diagnostic and therapeutic benefits of combined dermatology–rheumatology clinics.15 16 Generally, the focus of these clinics is psoriasis and PsA. To the best of our knowledge, no experience with combined clinics including other multidisciplinary professionals such as psychologists, social workers, dieticians and a broader rheumatology–dermatology–gastroenterology approach has been studied.
The overall aim of this study is to determine the effectiveness of an interdisciplinary combined clinic intervention compared with usual care in a population of patients with complex IMIDs, defined as more than one of the following diagnoses: psoriasis, HS, axSpA including AS, PsA, UC and CD. Our hypothesis is that an interdisciplinary combined clinic intervention will be more effective than usual care in improving patient-reported outcome (PRO) measures (ie, PROMs, including generic and disease-specific functional status, HRQoL, symptom and symptom burden and health-related behaviours) and clinical outcomes and that a more effective screening and management of other autoimmune diseases and comorbidities can be performed in an interdisciplinary combined clinic.
Methods
Trial design and setting
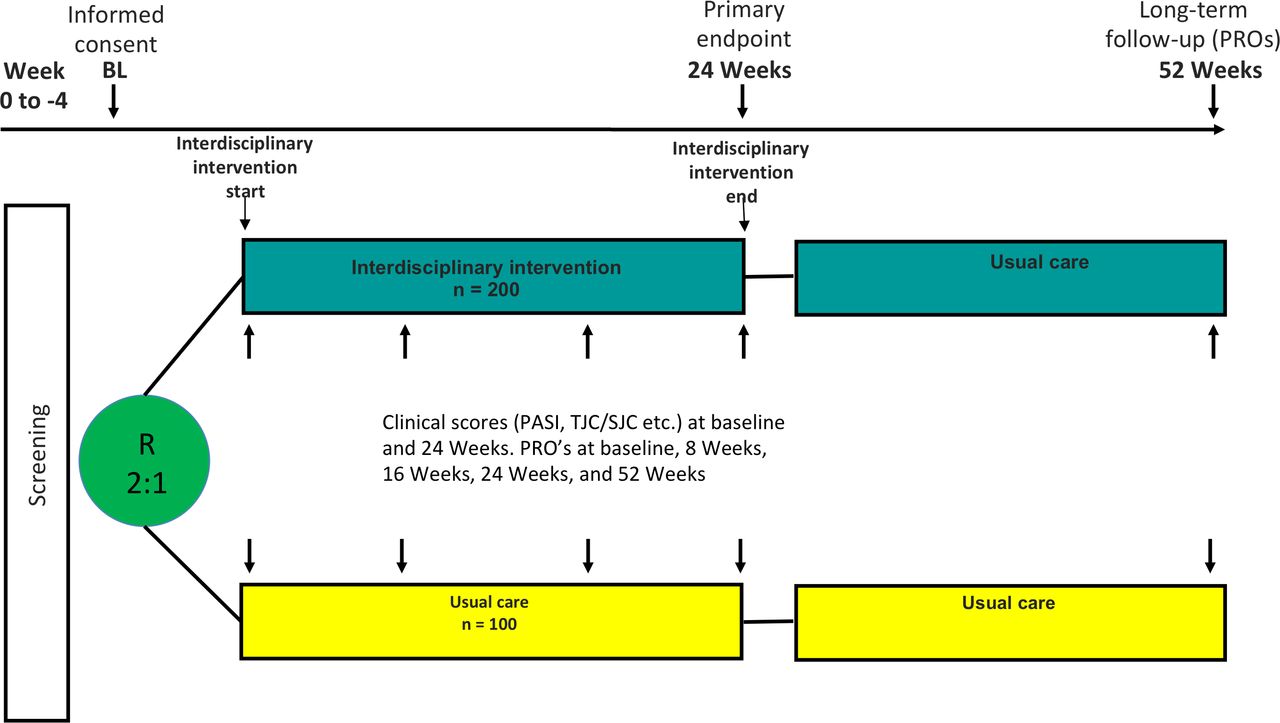
This is a randomised, usual care controlled, parallel-group clinical trial. Participants are enrolled consecutively and randomly assigned in a 2:1 ratio to either treatment in an interdisciplinary combined clinic or usual care in a hospital clinical setting. In total, 300 patients diagnosed with more than one of the selected IMIDs will be randomised to either interdisciplinary combined clinic intervention (200 subjects) or usual care (100 subjects). Work-up and therapy will be at the investigator’s/responsible physician’s discretion and in accordance with local and national treatment recommendations and guidelines. Thus, diagnostic procedures and therapy are not mandated by the study protocol.
Participants will be recruited based on referrals from hospital clinics and from consultative private practices.
The study will consist of a 6-month active intervention period (assessed after 24 weeks) and a subsequent 6-month follow-up period where no intervention or incentives will be provided by the trial. PROMs will be collected at baseline, 8, 16 and 24 weeks, as well as 52 weeks. Clinical endpoints will be collected at baseline and 24 weeks.
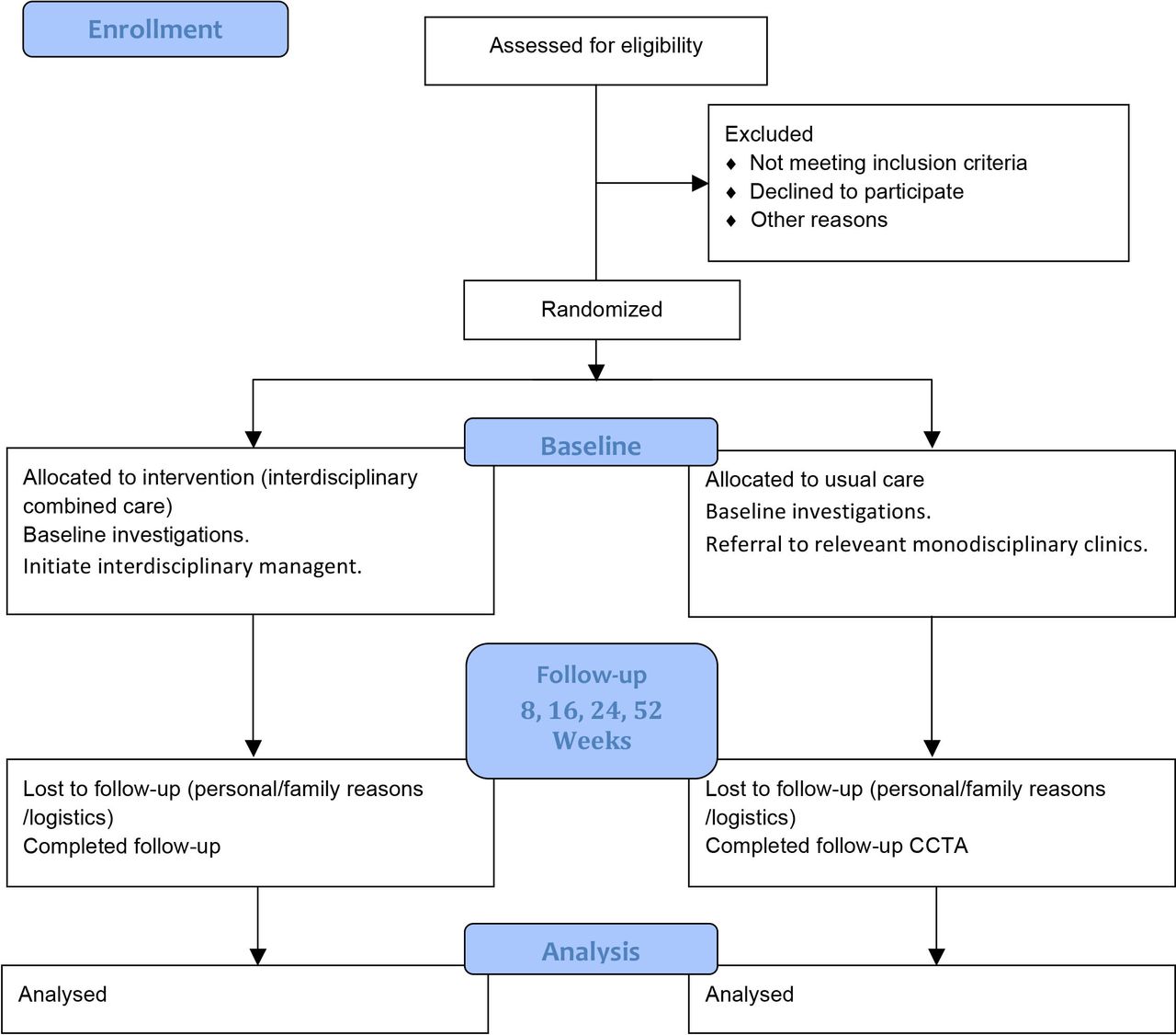
Figure 1 illustrates the study design. Figure 2 illustrates the trial flow.

Trial design. Two-arm, randomised, usual care controlled, parallel-group pragmatic clinical trial.

{kind=link}
{kind=link}
Study flow diagram.
Patient and public involvement
Two patient organisations (‘De Autoimmune’ and ‘Foreningen for Autoimmune Sygdomme’) were part of the original grant proposal, which formed the basis for establishing the National Centre for Autoimmune Diseases (NCAS). The trial described in this protocol is running in the NCAS. Members of the patient organisations provided feedback and comments on the trial concept. Other patients not directly associated with the patient organisations are providing feedback on the content of the interdisciplinary intervention throughout the trial. This feedback is organised through semistructured interviews and focus groups. Information about the trial is shared with patients through regional and national branches of the aforementioned patient organisations.
Record keeping, monitoring and data handling
Study data are collected and managed using REDCap (Research Electronic Data Capture) electronic data capture tools hosted at Aarhus University.17 18 REDCap is a secure, web-based software platform designed to support data capture for research studies, providing (1) an intuitive interface for validated data capture; (2) audit trails for tracking data manipulation and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages and (4) procedures for data integration and interoperability with external sources.
Personal data are protected according to the Danish Data Protection Act and The General Data Protection Regulation.
PROM data are collected as surveys through REDCap. The system will send customised emails to participants. It is ensured that participants can complete each survey one time only. Configurable reminders and tracking of responses are in place to minimise the risk of missing data. PRO results are available to investigators on an individual level as a tool to improve the treatment and the consultation. Data will not be available on trial level until database lock.
The Good Clinical Practice (GCP) unit at Aarhus University Hospital is granted access to perform monitoring to confirm that the trial is being conducted in accordance with the currently approved protocol and any other study agreements, International Conference on Harmonisation (ICH) GCP, and all applicable regulatory requirements.
Participants
Inclusion criteria
Written informed consent obtained from the participant prior to randomisation.
Age 18 and above.
Diagnosis of at least two IMIDs* or diagnosis of one IMID and clinical suspicion** of another IMID*
*Including and limited to psoriasis, HS, UC, CD, axSpA/AS and PsA.
**Substantiated by, for example, clinical findings, imaging, biochemical results or histological examination at the discretion of the investigator.
Exclusion criteria
Non-Danish speaking.
Expected to be unable to comply with the study protocol.
Recruitment and informed consent procedures
Participants will be recruited from the Department of Dermatology, Department of Rheumatology and Department of Hepatology and Gastroenterology, Aarhus University Hospital. Participants will also be recruited based on referrals from other hospital clinics and from consultative private practice.
Referred patients will be discussed at an interdisciplinary preadmission assessment. Patients who are potentially eligible to take part in the trial are invited to attend a clinic appointment. Potential participants will receive verbal and written information regarding the study. Participants will be offered the possibility for bringing a lay representative and will be offered time for reflection to decide whether they wish to participate in the study.
Randomisation and allocation concealment
Eligible participants will be randomised in a 2:1 ratio to either treatment in the interdisciplinary combined clinic or usual care. Participants are randomised by the investigator using a validated REDCap randomisation module. The sequence generation is based on computer-generated random numbers and created by the Clinical Trial Unit at Aarhus University using permuted blocks and no stratification.19 The investigators are blinded to the allocation sequence.
This is an open-label study and therefore both participants and investigators will be aware of allocation following the first enrolment visit.
Intervention
Interdisciplinary
The intervention in this trial consists of the combined efforts of the interdisciplinary team in the combined clinic arm. The intervention lies in the interdisciplinary organisation of workup, treatment and care for patients with complex IMIDs.
The interdisciplinary team consists of dermatologists, gastroenterologists, rheumatologists, nurses, psychologists, dieticians, social workers and secretaries. Physiotherapists are involved as needed. Treatment will be individualised based on clinical, biomarker, phenotypic and psychosocial characteristics. Consultations will be interdisciplinary and coordinated across disciplines. The medical treatment will follow local, national and international guidelines. Thus, the intervention is not a specific pharmaceutical treatment.
See online supplementary file for a detailed description of the intervention.
Usual care
Usual care will be carried out by HCPs who are not otherwise involved in the trial. In usual care, the patients will not be offered interdisciplinary patient-centred care as described, but rather attend their multiple usual disease-specific departments at the usual appointments. As participants will have complex IMIDs, this will typically entail attending multiple monodisciplinary specialised clinics. As in the interdisciplinary arm, treatment will be prescribed according to local, national and international guidelines by the treating physicians with no set protocol and no restrictions.
Trial objectives and endpoints
All primary and secondary objectives and endpoints are listed in table 1
Objectives and endpoints
Trial schedule and assessments
The study schedule (table 2) details the procedures and tests occurring at specific times throughout the study. Scheduled visits mandated by the protocol are for the purpose of data collection. Additional visits for workup, treatment and care will be scheduled individually based on the discretion of the treating team in both arms with no restrictions set by the protocol.
Study schedule
Adverse events
The objective of this study is effectiveness and not risk. Medicines are used in accordance with market authorisations and no specific medicines are being examined. The protocol does not endorse any prespecified treatment; rather medicines will be used at the physician’s discretion in both arms of the study. This trial does not fall under the definition of a clinical trial of medicinal products. Thus, suspected adverse drug reaction to medicines used in the trial will be subject to standard reporting to the Danish Medicines Agency according to standard clinical practice.
Reporting of suspected side effects from medicines are pursuant to the Danish executive order no. 381 of 9 April 2014 on the reporting of side effects from medicines, etc.
Serious Adverse Events1 (SAE)’s will be collected systematically in the trial at week 24 and if spontaneously reported from baseline to week 24. Drug relatedness of SAEs will be assessed by a trained physician. SAEs will be recorded in the medical record and the eCRF.
1An SAE is any untoward medical occurrence that
Results in death.
Is life-threatening.
Requires inpatient hospitalisation or prolongation of existing hospitalisation. (Planned hospitalisation or planned prolonged hospitalisation do not fulfil the criteria for being an SAE.)
Results in persistent or significant disability/incapacity.
Is a congenital anomaly/birth defect.
Is a medically important condition. Events that may not be immediately life threatening or result in death or hospitalisation but may jeopardise the subject or may require intervention to prevent one of the other outcomes listed in the definition above.
Sample size
The primary outcome is change in the physical component of HRQoL, measured using SF36 PCS, 24 weeks after randomisation.
Specification of the sample size calculation, including the target difference, is reported according to the guidance for reporting items available from the DELTA2 guidance on choosing the target difference and undertaking and reporting the sample size calculation.20 The assumptions regarding variation and expected effect assessed by changes in SF-36 are largely based on experience from previous pharmaceutical trials using SF-36 as a secondary outcome measure. Also, we have based our assumptions on Minimal Clinical Important Difference estimations from previous publications.21–23
The sample size of 300 patients (randomised: 200-to-100) is designed to provide a high statistical power (>90%) to detect a 5-unit difference in SF36-PCS change between the groups. All power and sample size calculations were conducted using R software V.3.4.3 (The R Foundation for Statistical Computing).
SF36 PCS: for a two-sample pooled t-test of a normal mean difference with a two-sided significance level of 0.05 (p<0.05), assuming a common SD of 10 SF36 points, a sample size of 85 patients per group has a power of 90% to detect a mean difference in the group mean changes of 5 SF36 points (corresponding to a moderate Cohen’s effect size of 0.5). Due to a very limited experience with attrition, to utilise the capacity of the clinic, to maximise data generation in the combined clinic arm and to increase external validity of the study, it was decided to aim for enrolment of 300 participants in total; with a majority (200 patients) being randomised to the interdisciplinary intervention. With 100 patients in each group in the intention-to-treat (ITT) population, the statistical power might be as high as 94% based on the assumptions above.
Statistical analysis
All p values and 95% CIs will be two sided. We will not apply explicit adjustments for multiplicity, rather we will analyse the key secondary outcomes in a prioritised order (eg, using ‘gatekeeping procedure’); that is, the analyses of the key secondary outcomes will be performed in sequence until one of the analyses fails to show the statistically significant difference, or until all analyses have been completed at a statistical significance level of 0.05.24 The key secondary statistical tests will be reported with p values for hypothesis tests and claims of statistical significance. The primary statistical model will consist of repeated-measures linear mixed models to compare patient outcomes trajectory over time between the two intervention groups (ie, Time×Group interaction).
The prespecified analyses will be based on the ITT population, using data from the full-analysis set, which will include all patients who underwent randomisation and had at least the outcome of interest measured at baseline.25 Data will be analysed using R and SAS or STATA, with the particular outcome variable at baseline level as a covariate—using a multilevel repeated measures mixed effects model with participants as the random effect factor based on a restricted maximum likelihood model. The primary outcome analyses for continuous outcomes will be based on the following model: the dependent variable (eg, change in the SF36 PCS value) will be the response variable, and the baseline value (one for each participant) will be applied as a covariate, with a fixed effect (main effect) for treatment group (two levels), IMID condition (five levels; corresponding to psoriasis, HS, PsA, axial spondyloarthritis and IBD) and time (four levels: 0, 8, 16 and 24 weeks) will be included as covariates, as well as the interaction between treatment group and time (Group×Time), and Patient ID as a random effects factor. This statistical model will hold all between-group comparisons at all assessment points (incl. baseline) and allows for evaluation of the average effect, as well as the trajectory over the time period from baseline to 24 Weeks follow-up.26 Results will be reported as the difference between least squares means and their corresponding 95% CI.
Categorical changes for dichotomous end points will be analysed with the use of logistic regression with the same fixed effects and covariates as the respective analysis of continuous outcomes; ORs (and 95% CI) will subsequently be converted into risk ratios (RRs, and 95% CI).
Handling of missing data and sensitivity analyses
We plan to conduct both an analysis of the full analysis set (ITT population) and a per protocol analysis, so that any differences between them can be explicitly discussed and interpreted. Using mixed models, like described above, provide valid estimates of treatment effects even when the missing values are not completely random,26 and additional methods for handling missing data, such as multiple imputation, are generally not required.
Missing data will be handled by:
Attempt to follow-up all randomised participants, even if they withdraw from allocated treatment.
Perform a main analysis of all observed data that are valid under a plausible assumption about the missing data (ie, model-based: data as observed; using linear mixed models assumes that data are ‘Missing At Random’ (MAR).
Perform sensitivity analyses to explore the effect of departures from the assumption made in the main analysis (ie, a non-responder-imputation: using the value at baseline to replace missing data will correspond to a non-responder imputation; these models will potentially be valid even if data are ‘Missing Not At Random’ (MNAR).
Account for all randomised participants, at least in the sensitivity analyses (covered by #2 and #3 above plus the corresponding analyses based on the Per protocol population).
The interpretation of the corresponding statistical measures of uncertainty of the treatment effect and treatment comparisons will involve consideration of the potential contribution of bias to the p value, 95% CI and inference in general.
Our primary analysis population will be all participants with available data at baseline statistically modelled using repeated-measures linear mixed models (see above). These models will be valid if data are MAR.
#3+4 Sensitivity: We will analyse all variables with missing data being replaced by imputation of the baseline level; that is, interpreted as assuming that those who dropped out returned to their baseline level; these estimates could potentially be valid even if data are MNAR.
Ethics and dissemination
The risks and burden associated with participating in this clinical trial are considered low and outweighed by the benefit of achieving high-quality scientific knowledge regarding the potential benefits of treating patients with complex IMIDs in an interdisciplinary combined clinic setting. Additionally, on the individual level, participants are expected to experience immediate diagnostic and therapeutic benefit from the interdisciplinary approach. Ethical approval of this study protocol was established by the Central Denmark Region Ethical Committee. The findings from this trial will be disseminated via conference presentations and publications in peer-reviewed journals, and by engagement with patient organisations.
Discussion
For the purpose of the current trial, a number of prototypical IMIDs have been chosen: psoriasis, HS, UC, CD, axSpA and AS and PsA. These diseases will serve as a model for autoimmune diseases in which an interdisciplinary and combined clinical approach will be tested. We believe the model will be scalable with the potential to include other IMIDs in the future.
This study has the potential to address some of the main challenges for IMIDs regarding the management of the complexity of the diseases and comorbidities. The focus of the study will be on personalised, preventive and participatory healthcare.
As described above, patients often have more than one IMID, which lead to patients often need to attend several departments. Patients report communication problems between the departments, experience of neglect regarding comorbidities and that they are left with the responsibility for coordinating the different treatment courses between the different departments.12–14
An increasing body of literature supports that IMIDs share many immunopathogenic features and that there is a considerable clinical and therapeutic overlap between the diseases.1 27 28 This underlines the need to abandon previous perceptions of IMIDs as based on cluster of symptoms and a specific silo in the healthcare system. Rather, IMIDs must be seen as chronic conditions that may affect a number of body functions and other patient-relevant social and personal aspects. This calls for an integrated and interdisciplinary approach, which will be in scope for this study. Previous efforts to improve patient-centricity within IMID’s through combined clinics have typically included only two medical specialties, for example, rheumatology and dermatology.15 16 The novelty of our concept is first that it includes a broader range of relevant medical specialties spanning a range of inflammatory diseases affecting the skin, musculoskeletal system and gut. Second, the concept adheres to a holistic treatment approach, as other cross-disciplinary professionals are part of the team. Third, the effectiveness of the interdisciplinary combined clinic approach is assessed through data generation in a randomised, usual-care controlled trial setting which has not previously been done.
If it is shown that an interdisciplinary patient-centred approach improves quality of life in these patients compared with usual healthcare, professionals may rethink the way the health system is organised and ultimately implement an interdisciplinary approach in the management of IMIDs.
Another aspect that will be explored in this project is whether an interdisciplinary patient-centred approach is associated with a socioeconomic benefit, for example, by reducing patients’ sick leave, need for attending to healthcare and lower medicine costs.
There is currently a political and patient-driven move towards an interdisciplinary treatment approach. However, for this to be broadly generalisable, the potential advantages must be proven towards the usual and traditional therapeutic approach.
The pragmatic elements in the design of this trial increase the likelihood that the results can be generalised to everyday practice and support decision-making by patients, providers and health system leaders. The use of a generic PRO as the primary outcome is remarkable and creates a strong focus on patient-centricity. A generic PRO that can be used across age, disease and treatment groups enables a meaningful assessment of patients with complex IMIDs.29–31
However, there are some limitations in this study. The minimisation of inclusion and exclusion criteria, the potential diversity of individualised treatments, and participants’ experience and expectancy of living with a chronic disease may introduce additional variables, which may affect the outcomes. The 24 weeks duration of the intervention may be insufficient to provide the full benefit in the selected group of patients with chronic, long-standing, complex IMIDs and comorbidities. Sample size calculation is based on the primary outcome, change in SF-36 PCS, whereas the trial may be underpowered to assess changes in subgroups of participants within each disease domain. Thus, there may be insufficient statistical power to determine the effect of the intervention on certain secondary endpoints.
Furthermore, investigators and patients cannot be blinded to participation randomisation outcomes due to pragmatic design limitations. Increased disease awareness in the usual care group caused by participating in the trial may potentially reduce the difference between the intervention group and the usual care group.
A potential bias may be introduced as patients might be inclined to report improvements in generic and disease-related PROs based simply on the fact that they have been assigned to one study arm or the other. However, findings from published psychobehavioral literature suggest that cognitively, respondents are not prone to altering the content of their self-reports of symptoms associated with treatments that they are receiving,32 and an analysis of the trustworthiness of PROs in unblinded cancer clinical trials did not find evidence of a bias associated with knowledge of treatment allocation.33 Furthermore, patients in this study is not assigned to placebo but will receive medical care no matter of trial arm allocation. In fact, patients in the usual care arm may likely improve due to medical treatment decisions as they will likely by referred to the trial in a period with disease activity and thus indications for treatment modifications.
Nonetheless, the results and experience from this study may reveal the benefits of managing patients with complex IMIDs in an interdisciplinary setting. The trial may provide evidence as to whether an interdisciplinary approach to complex autoimmune diseases is beneficial for the patients and lower the socioeconomic burden.
This could form the basis for establishing further interdisciplinary autoimmune clinics on a national and international scale.
Trial status
This trial is ongoing. The first participant was enrolled on 14 January 2020.
Supplemental material
Acknowledgments
The authors wish to acknowledge the entire team in the National Center for Autoimmune Diseases for their dedicated work; including current and previous team members: Lise Guld Lerke-Møller, Rikke Edelbo, Mia Marie Remmer, Anja Astrup, Caroline Vinther Hammelsvang, Karen Margrethe Schifter Kirketerp, Helene Almind Pedersen, Louise Gude Jensen, Inger Nielsen Larsen. Sanne Schou. The authors also wish to acknowledge the patient organisations ‘De Autoimmune’ and ‘Foreningen for Autoimmune Sygdomme’ for collaboration in establishing the National Center for Autoimmune Diseases.Professor Christensen wants to acknowledge that the Parker Institute, Bispebjerg and Frederiksberg Hospital is supported by a core grant from the Oak Foundation (OCAY-18-774-OFIL).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors KFH is the principal investigator and is responsible for leading the design phase and drafting of the protocol. All authors (KFH, AD, JA, TBL, AGL, LFM, RC, LI) made contributions to the design of the trial and have been involved in drafting the manuscript or revising it critically for important intellectual content. All authors (KFH, AD, JA, TBL, AGL, LFM, RC, LI) read and approved the final manuscript. LI wrote the project grant application and was awarded funding to establish the centre in which the trial is being run.
Funding The basic costs of establising and running the interdisciplinary clinical centre (Nationalt Center for Autoimmune Sygdomme) are supported by a grant from The Danish Ministry of Health. Award/grant number not applicable.
Competing interests AD has received speaking fees from Pfizer. AGL has been a consultant and advisor for the following companies: AbbVie, Eli Lilly, MSD, Novartis, Pfizer and UCB and has received speaking fees from: AbbVie, MSD, Novartis, Pfizer and UCB. JA has been consultant, advisory board member or speaker for the following companies: AbbVie, MSD, Bristol Meyer Squibb, Ferring Pharmaceuticals, Pfizer, Janssen-Cilag and Takeda. KFH has been a consultant and advisor for the following companies: AbbVie, LEO Pharma, Novartis and has received speaking fees from: AbbVie, LEO Pharma, Novartis, Janssen, CSL Behring. LI has served as a consultant and/or paid speaker for and/or participated in clinical trials sponsored by: AbbVie, Almirall, Amgen, Astra Zeneca, BMS, Boehringer Ingelheim, Celgene, Centocor, Eli Lilly, Janssen Cilag, Kyowa, Leo Pharma, MSD, Novartis, Pfizer, Samsung, UCB. LFMø has been advisory board member for Janssen and has received speaking fees from LEO Pharma. Robin Christensen reports no conflicts of interest. TBL has been a consultant and advisor for UCB.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.