Article Text

Abstract
Introduction Compression is the mainstay of treatment for venous leg ulcers (VLUs) and there are few effective adjuvant treatments. There is only observational evidence supporting the use of hypochlorous acid (HOCl) as a topical wound solution on VLU and some limited randomised evidence for the effect of a prescribed regimen of exercise.
Methods and analysis The Factorial4VLU trial is a pragmatic, blinded, factorial randomised controlled trial, with 380 participants receiving either a prescribed exercise regimen compared with usual care and either active HOCl wound solution or placebo wound solution at each dressing change for up to 24 weeks. All participants will receive compression therapy. The primary outcome is the proportion of participants with healed VLU at 12 weeks after randomisation as adjudicated by blinded review of ulcer photographs. Secondary outcomes are proportion healed at 24 weeks, time to healing, estimated change in ulcer area, change in 2-Minute Walk Test, change in health-related quality of life, incidence of infection and incidence of all-cause adverse events. If either of the interventions shows a statistically significant positive difference on healing outcomes, cost-effectiveness will be modelled using a health service perspective.
Ethics and dissemination The Factorial4VLU trial received ethical approval from the Northern B Health and Disability Ethics Committee. We plan to publish the results within 1 year of trial completion and will include the results on the trial registration page.
Trial registration numbers Australia and New Zealand Clinical Trials Register (http://www.anzctr.org.au) (ACTRN12620000116921); Universal Trial Number (WHO) (U1111-1236-2997).
- wound management
- primary care
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This trial will evaluate the effectiveness of two interventions within one trial.
This trial uses a pragmatic design to maximise external validity of the findings.
While the hypochlorous acid intervention is fully blinded, the exercise intervention is single blinded with blinded adjudication of the complete healing outcomes via ulcer photographs. Being open label for all other outcomes in the exercise arm may introduce bias into some outcome assessments, for example, health-related quality of life and incidence of adverse events.
Introduction
Effective treatment for venous leg ulceration (VLU) involves correcting the underlying venous hypertension associated with chronic venous insufficiency with a form of compression therapy. However, about half of treated patients remain unhealed after 12 weeks.1 2 Early endovenous ablation of superficial venous reflux increases VLU healing,3 but the procedure is not funded in New Zealand and may not be readily available or accessible in other jurisdictions. Pentoxifylline is available in New Zealand,4 but its use is limited, despite guideline recommendations.5 Thus, other adjuvant treatments are needed to promote ulcer healing. In this study, we examine the role of exercise and topical hypochlorous acid (HOCl) as adjuvant treatments.
Exercise and VLU healing
Calf muscle contraction promotes venous return but dysfunction in the calf muscle pump along with valve impairment or damage in the venous bed leads to venous hypertension allowing fluid and large molecules to escape into surrounding tissue, causing oedema and trophic skin changes. The most severe effect of these changes is VLU. VLU is associated with poorer calf muscle strength and reduced calf muscle endurance,6 7 lower calf muscle pump ejection fractions and greater residual volume fractions.8 Patients with VLU also tend to walk less than those without VLU.9
A systematic review of adjuvant exercise interventions for treating VLU found five trials (n=190) of different exercise regimens that could be included in a meta-analysis.10 The overall effect was in favour of exercise, with 14 extra cases healed per 100 patients at 12 weeks (risk difference 14%, 95% CI 1% to 27%). Subgroup analysis by type of exercise regimen showed the addition of 30-minute walking three times per week, or a similar activity, to a daily regimen of progressive resistance exercise appeared to increase VLU healing at 12 weeks by an extra 27 cases healed per 100 patients (two trials, n=102, risk difference 27%, 95% CI 9% to 45%).
The two trials in this subgroup were small and differed in mode of delivery. One trial (n=63) was home based, while the second trial was gym based and there were differences in the exercise regimens.11–13 However, the use of gyms is not a scalable intervention in most countries, whereas home-based exercise regimens that do not require extra equipment are an intervention that could be widely deployed.
The effects of HOCl on wound healing
Static progress to wound healing is associated with chronic infection and may be caused by biofilms.14 However, identifying biofilm in wounds is difficult as wounds with devitalised tissue do not invariably support a biofilm, whereas wounds that appear healthy can support biofilm.15 Bioburden (the quantity of bacteria present in a wound) is also associated with wound duration, with longer duration wounds having a greater presence of bacterial species that are known to inhibit healing.16
HOCl is water and sodium chloride electrolysed to create reactive oxygen and chlorine in a pH neutral solution that has antimicrobial activity.17 HOCl can disrupt and prevent biofilm and is bactericidal in low concentrations against common species that are associated with delayed healing, for example, Pseudomonas spp.17 Two trials (n=100) of HOCl in diabetic foot ulcers (DFUs) have used healing as an outcome, but both trials compared HOCl with povidone-iodine, which is not a suitable comparison group to extrapolate to VLU.18 19 HOCl use has been reported in four case series of VLU, with minimal adverse effects and suggested improvements in healing,16 17 20 21 but there are no randomised trials of HOCl in VLU.
Thus, there is a need for definitive trials of both exercise and HOCl for treating VLU. The aim of this factorial trial is to evaluate the effectiveness of each intervention on VLU healing as an adjunct to compression therapy.
Methods and analysis
Trial design
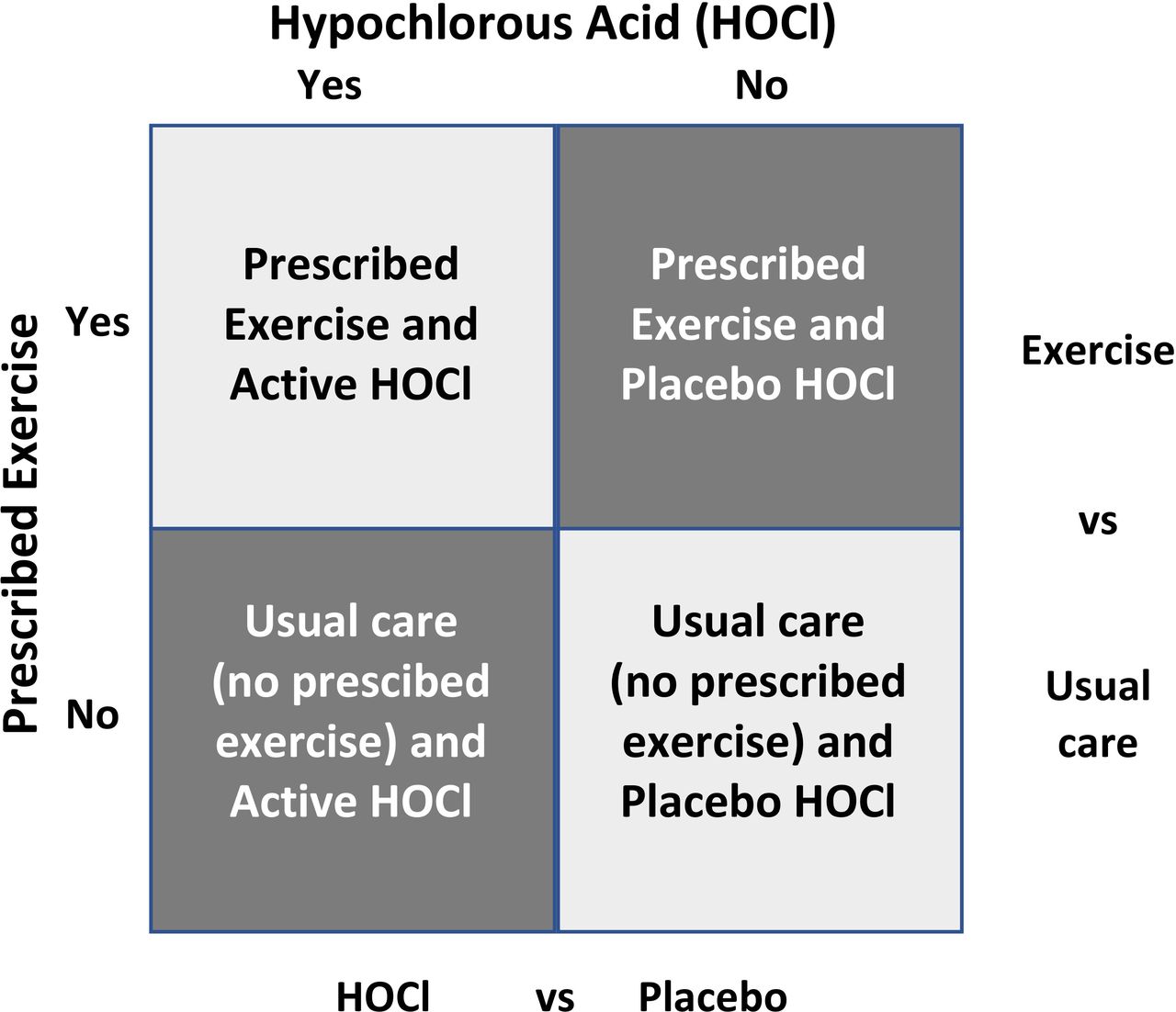
The Factorial4VLU trial is a pragmatic, blinded, factorial randomised controlled trial with participants receiving either a prescribed exercise regimen compared with usual care and either HOCl wound solution or placebo wound solution at each dressing change (figure 1) for up to 24 weeks. Block randomisation is being used, stratified by study centre and prognostic index (ulcer size and duration)22 to ensure a balance of participants with severity of ulceration within study centres and to isolate any centre effect. Participants in both arms will receive compression therapy (specific compression system guided by patient and/or clinical preference), which is standard care delivered by district nursing services at the study centres.

Factorial4VLU trial design showing each allocation by cell.
Case definition
A patient will be considered to have a VLU where other causative aetiologies have been ruled out, the ulcer appears clinically venous (presentation may include any or all of the following: moist, shallow, irregular shape; haemosiderin pigmentation; venous eczema; ankle oedema; ankle flare; lipodermatosclerosis), and the patient has an Ankle Brachial Index of greater than or equal to 0.8 obtained within the last 3 months to rule out significant arterial insufficiency. There will be no minimum or maximum ulcer area for inclusion.
An incident VLU will be either (a) any new lesion in the skin on the leg (below the knee) that has been present for 4 or more weeks and meets the case definition for VLU, or (b) where a patient with a history of VLU re-presents to the service earlier than 4 weeks since onset of the lesion where the new ulcer is diagnosed as a VLU. Patients already in compression treatment for VLU will be considered to have prevalent VLU. Patients with incident and prevalent VLU will be eligible for the trial.
Inclusion criteria
Patients with leg ulcers will be eligible for inclusion if they are aged 18 years or older and able to provide written informed consent, determined to have a VLU, able to tolerate compression therapy and are judged as being able to safely mobilise (with or without walking aids) by the research nurse.
Exclusion criteria
Patients will be excluded from participation if they have a hypersensitivity to HOCl or sodium hypochlorite, have a fixed ankle joint (ie, surgically fixated, not a joint with restricted range of motion), a VLU with exposed bone or tendon, a history of rheumatoid arthritis or vasculitis, uncontrolled diabetes (defined as glycosylated haemoglobin >100), severe liver failure (indicated by jaundice), severe heart failure (defined as short of breath while seated), or renal failure (estimated glomerular filtration rate <30), severe peripheral arterial disease (defined by inability to walk even short distances without pain), suspected or diagnosed skin malignancy associated with leg ulcer, or any other threat to safe participation. Patients with wound infection requiring treatment with antibiotics or a medicated dressing at registration will be deferred from consideration until the infection resolves.
Consent
All participants will receive a participant information sheet (PIS) that includes the informed consent form (ICF). The research nurses will discuss the PIS/ICF with potential participants as well as any other persons the participant wishes to include, for example, caregivers or family members. Participants must give written informed consent if they wish to participate. Participation is voluntary and participants will be free to withdraw at any time without this decision influencing their usual treatment.
Recruitment
Patients with VLU will be recruited from the community-based district nursing services at study centres, starting 1 March 2020 through to 30 October 2021, with follow-up concluded by 1 May 2022. The centres are based in the Auckland, Counties Manukau, Capital and Coast, and Southern District Health Boards district nursing services and the Nurse Maude Association community nursing services in four cities in New Zealand (Auckland, Wellington, Christchurch and Dunedin). A 0.5 full time equivalent research nurse has been seconded to the trial from each study centre. All but one research nurse have been involved in our previous trials and they all received 2-day induction training in the trial procedures, which has been followed up by six weekly Zoom meetings to discuss and resolve any implementation issues.
Potentially eligible patients will be identified from screening by the research nurses or by notification from community nurses (figure 2). The research nurses will phone and visit these patients to screen for eligibility and obtain informed consent to participate. The location for these visits may be a clinic, the patient’s home, workplace, or other venue convenient to, and nominated by, the participants. Eligible patients with incident VLU must be treated for 1 week with compression therapy before randomisation to ensure they are tolerant of compression. Patients with prevalent VLU in compression may be randomised immediately after registration and baseline assessments.

{kind=link}
{kind=link}
Detailed trial schematic showing participant contacts and measures. HRQoL, health-related quality of life; ICF, informed consent form; PIS, participant information sheet; VLU, venous leg ulcer.
Randomisation and allocation concealment
Participants will be randomised in a 1:1 ratio to one of the four trial cells using stratified block randomisation with block sizes of 4. Randomisation will be stratified by trial centre and prognostic index (one stratum being VLU <5 cm2 and <6-month duration with the second stratum being either VLU area >5 cm2 or VLU duration >6 months, or both VLU area >5 cm2 and VLU duration >6 months).22 The randomisation sequence will be prepared by the trial statistician and loaded into Research Electronic Data Capture (REDCap) databases to be accessed one participant at a time by the research nurse via a computer tablet (Samsung S6 LTE) at the point of randomisation.
Blinding
The HOCl wound solution arm of the trial will be placebo-controlled (purified water with added preservative to provide the same shelf life once opened as the HOCl solution) with the active and placebo solutions contained in matching bottles. The investigators, participants, research nurses and district nurses will be blinded to the intervention in the HOCl arm of the trial.
It is not possible to blind participants, research nurses and community nurses to the participant’s assignment to exercise. However, outcome assessment for the primary endpoint and two secondary endpoints (proportion healed at 24 weeks and time-to-complete healing) will be blinded by using assessors (AJ, JW) unaware of treatment allocation to adjudicate the healing status of the ulcer from the endpoint photographs. Where a status cannot be determined from the photograph, the ulcer will be considered unhealed. Where the adjudicators disagree, they will meet to arrive at a consensus decision on the ulcer status. These processes were successfully used in a previous trial, where the agreement between the adjudicators was 91% (kappa 0.81), and agreement between the research nurses and adjudicators was 96% (kappa 0.93).23
Photographs of the leg and reference ulcer will be taken at baseline and endpoint by the research nurses using a Panasonic Lumix TZ80 with autoflash and macro-settings and loaded onto a secure web-based server (OwnCloud). The baseline photographs will be available to the adjudicators to ensure the same ulcer (or ulcer site) is being reviewed for the 12-week and 24-week endpoint adjudications. The photographs will be assessed by the study project manager and the principal investigator shortly after each visit to ensure image quality. If the image quality is poor, the research nurse will return to the participant to rephotograph the leg and reference ulcer (or ulcer site).
On study completion, the Trial Steering Committee (TSC) will interpret study results blinded to treatment allocation and the code broken only after TSC agrees to an interpretation.
Sample size calculation
We estimate 380 participants will be required. A sample size of 344 will be sufficient to show a 17% absolute difference in proportion completely healed at 90% power with an alpha of 0.05 at 12 weeks. This difference is smaller than the 27% difference in healing found in the systematic review of exercise trials or the 55% difference in healing found in the HOCl trial in DFUs.10 18 Despite our previous trials having very high follow-up rates (98%–100%) on the primary outcome,1 2 24 we have allowed for 10% loss to follow-up, and thus 190 in each group will be required if our assumptions are correct.
While a minimum of 24-week follow-up is recommended in VLU trials and a time-to-event outcome is preferable for primary outcome,25 we had no information on the likely effects of either exercise or HOCl on VLU healing at 24 weeks on which to base a sample size calculation. We did have sufficient information to use for number healed at 12 weeks and hence our choice of that outcome as primary outcome on which to base a sample size calculation.
Baseline data collection
Demographic data collection will include age, sex, self-identified ethnicity, education, employment, height, weight, smoking history, ulcer history (duration, size, number of episodes, type of compression system, Clinical Venous Severity Score and pain), clinical history (diabetes, history of deep vein thrombosis, joint replacement, leg fractures, treatments for varicosities), estimated walking time per week and current medications (table 1). We will also collect health-related quality of life (HRQoL) assessments and distance walked using a 2-Minute Walk Test (2MWT). All data will be entered into REDCap by the research nurses.
SPIRIT schedule of of enrolment, interventions and assessments
Primary outcome
The primary outcome is the proportion of patients to have a healed reference ulcer (defined at baseline assessment as the largest VLU where there is more than one VLU) at 12 weeks as adjudicated by the blinded review of ulcer photographs. Healing will be defined as complete epithelialisation of the reference ulcer with absence of scab. If a light scab is present, the research nurses will gently remove it in order to determine whether there is complete epithelialisation under the scab.
Secondary outcomes
Secondary outcomes are limited to time to healing, proportion of patients with a healed VLU at 24 weeks, change in ulcer pain to 12 weeks, estimated change in ulcer area from baseline to 24 weeks, change in 2MWT at 24 weeks, change in HRQoL at 24 weeks, exercise adherence to 24 weeks, and incidence of systemic VLU infection and incidence of adverse events (table 1). Agreement between the research nurse assessors and the blinded adjudicators on the 12-week and 24-week healing outcomes will be reported. While high levels of agreement have been reported in trials reporting both blinded and open-label outcome assessor, unblinded assessors do overestimate treatment effects.26
Change in ulcer area will be estimated using a pragmatic approach to ulcer measurement. We will use maximum ulcer width and length as inputs into calculating the area of an ellipse. The area of an ellipse is closely correlated to actual ulcer area (r=0.95),27 which we have verified from analysis of our data (r=0.92) from a previous trial.1
Change in HRQoL will be assessed using two generic instruments, the Short Form-12 and EuroQoL 5 Dimension 5 Level (EQ5D-5L),28 29 and one disease-specific instrument, the Charing Cross Venous Ulcer Questionnaire.30
For change in ulcer pain, all participants will self-record ulcer pain once each week for 12 weeks using a 0–10 pain score in a participant diary, prompted by an automated email reminder or a text from the research nurse (where the participant does not have an email address).
The 2MWT is an objective measure of function. It will be measured at baseline and at 12 and 24 weeks. Between-test changes of about 12 m on the 2MWT are considered a meaningful clinical difference.31 Change in 2MWT will obtained from a timed test of all participants at baseline, 12 and 24 weeks. Each participant will be asked to walk around an indoor course of two cones laid out 3 m apart within the visit setting, that is, in a hallway within the home or clinic. The number of 3-metre lengths completed within 2 min will be counted and the completed distance estimated from the number of lengths. Where a participant finishes the 2 min between the cones, the distance will be measured from centre of the last cone passed to the heel of the participant.
Exercise adherence for the participants in the exercise arm will be measured until 24 weeks. Participants will be asked to log the completed number of heel raises and walking each day in an exercise diary provided at randomisation until the 24-week endpoint. Participants will be sent an automated email reminder each week (generated in REDCap), or if they do not have an email address, they will receive a weekly text or call from the research nurse.
Incidence of systemic VLU infection will be measured three ways: (1) proportion of patients that have suffered infections up to 24 weeks; (2) incidence of VLU infections up to 24 weeks and (3) time to first episode of infection at 24 weeks. We will use a simple empirical definition of infection, namely presence of signs and symptoms of infection in the ulcer combined with treatment with antibiotics. Episodes of infection will be treated as independent if more than 21 days passes between prescriptions. We will obtain the information about antibiotic use as part of concomitant medication data collected at 12 and 24 weeks, and from the participant’s clinical record and electronic community-dispensing portals.
All-cause adverse events, including serious adverse events (SAEs), will be reported to the research nurse by either participants or district nurses throughout the follow-up period. Reporting will also be prompted during research contacts at weeks 12 and 24. An adverse event will be defined as any untoward medical event irrespective of whether it is thought to be related to the treatment or not. Adverse events reports will be reviewed by one of the medically qualified co-investigators blinded to participant allocation, with the relationship to treatment coded using the WHO Uppsala Monitoring Centre system for causality assessment.32 Adverse events will be coded using the Medical Dictionary for Regulatory Activities and study-specific leg ulcer codes. SAEs will include hospitalisation or prolongation of hospitalisation, life-threatening condition, significant disability or impairment, death, birth defect or any other important medical event. SAEs will be notified to the study project manager as soon as they are detected and will be monitored to resolution by the coordinating centre. Although not anticipated, any sudden unexpected serious adverse reactions (SUSARs) will be documented and notified to TSC, the Data Safety Monitoring Board (DSMB), the ethics committee, and the manufacturer, if appropriate, as soon as the coordinating centre is alerted to the SUSAR.
Cost-effectiveness modelling
Should either of the interventions be effective, we will conduct cost-effectiveness modelling to assess the costs and consequences of using exercise or HOCl as an adjuvant to compression therapy in community-based participants with VLUs. We will use a health service perspective to identify the cost of the interventions. We know from a previous study that the major cost drivers will be the use of health services.1 These costs include district nursing time, dressing materials and compression bandages used at each visit, general practitioner and other practitioner visits for VLU care, drugs dispensed for VLU treatment and secondary care (emergency department visits and days hospitalised for VLU treatment). We will be able to determine the number of community nursing visits, community drug dispensing and hospitalisations from electronic records at each study site. We will elicit information on compression systems and dressings at the baseline, 12-week and 24-week visits, as well information about other primary care visits during the 12-week and 24-week visits. We will use published resources to estimate costs, with local costs used if published resources are not available. We will establish health utilities using the EQ5D to produce quality-adjusted life years (QALYs). Costs will be annualised (as the follow-up period is for 24 weeks) and the final models (base case and alternatives) will report on the incremental cost-effectiveness ratio (cost per additional healed VLU) and cost per QALY. We will use bootstrapping to estimate levels of uncertainty around the point estimates.
Statistical analyses
Data analyses will be specified a priori in a statistical analysis plan prepared by the trial statistician (and agreed by all members of the TSC). Data will be entered into REDCap databases hosted at the University of Auckland. REDCap is a secure, web-based application designed to support data capture for research studies.33 The data will be extracted into SAS V.9.4 (SAS Institute) for analysis.
Analysis will be at the margins comparing each intervention (exercise vs no exercise, HOCl vs placebo). There will be no analysis for interaction and analyses will use the intention-to-treat principle. Binary outcomes will be analysed using logistic regression. Sensitivity analyses will be undertaken to determine the impact of missing data and adherence. Continuous outcomes (with 95% CIs) will be analysed using multiple linear regression and adjusted for baseline value and other covariates if needed. Change in ulcer-related pain will be analysed using repeated measures model with adjustment for baseline value. Time-to-event data will be analysed using Kaplan-Meier plots and log rank test. Cox regression will be used with time-to-event data adjusted for stratification factors and any imbalance in covariates. The assumption of proportionality will be checked using standard graphical techniques. Adverse events will be analysed using incidence rate ratios with reporting guided by the Consolidated Standards of Reporting Trials extension statement on reporting of harms and a recent systematic review on adverse event reporting in VLU trials.34 35 No interim analyses will be undertaken.
Should the treatments prove effective in the primary analysis, heterogeneity of effects will be analysed on proportion healed using subgroups specified a priori in the analysis plan. Likely subgroups will be age, sex, ethnicity, ulcer size at baseline, ulcer duration, prognostic index and study centre.
Treatment period and follow-up
The allocated treatment will be continued until complete healing or end of the trial after 24 weeks’ treatment. The research nurses are trained in leg ulcer care and most have been involved in previous 4VLU trials. The research nurses will deputise for the community nurses on research visits. In between research visits, care will be delivered by community nurses. All participants will be followed up at the scheduled timepoints irrespective of whether they continue with the assigned treatment (figure 2). The only exception to follow-up will be if the participant requests withdrawal of their data from the trial.
Intervention and control treatments
Participants will be allocated to one of four cells in this factorial trial (figure 1) and these treatments are all in addition to compression therapy.
Cell 1: prescribed regimen of exercise and active HOCl. The prescribed regimen of progressive resistance exercise consists of daily lower leg exercises to increase calf muscle strength at least until ulcer healing (three sets of seated exercises starting with 10 repetitions per set and increasing up to 25 repetitions per set, before progressing to standing two-legged exercises and then standing one-legged exercises) with the participants' body weight providing the resistance element. Participants progress to the next stage of exercise when they are comfortable with achieving the current step. The exercise regimen is supported with a resource book that provides general advice about venous ulceration and demonstrates the exercises, plus includes a goal setting diary (which will be assisted by trained research nurses) and an exercise diary to record adherence. The prescribed regimen also includes a target of walking for 30 minutes each day, 3 days per week. The regimen will be demonstrated one-to-one by the research nurses. Participants will perform the resistance exercises wherever they are comfortable, for example, their own home for the duration of the trial (24 weeks) or until the ulcer is healed, whichever is earlier.
The HOCl wound solution will be sprayed on the ulcer at each dressing change, the wound debrided as required, devitalised tissue and debris will be removed, and then the wound solution reapplied before the ulcer is redressed. Venous ulcers are typically redressed once per week, but may be redressed more frequently as indicated. Dressings are usually changed by nurses, but may be changed by some self-managing patients if they are wearing compression hosiery or wraps. The wound solution will be used for the duration of the trial (24 weeks) or until the ulcer is healed, whichever is earlier.
Cell 2: prescribed regimen of exercise and placebo HOCl (purified water plus preservative). The prescribed regimen of progressive resistance exercise and the use of the purified water wound solution is as described in arm 1.
Cell 3: usual care and active HOCl wound solution. The usual care regimen is supported by a participant resource book that provides general advice about venous ulceration. The wound solution will be sprayed on the ulcer at each dressing change, the wound debrided as required, devitalised tissue and debris will be removed, and the the wound solution reapplied before the ulcer is redressed. The wound solution will be used for the duration of the trial (24 weeks) or until the ulcer is healed, whichever is earlier.
Cell 4: usual care and placebo HOCl (purified water plus preservative). The usual care regimen and the use of the purified water wound solution will be as described in arm 3.
Background treatments
All participants will receive compression therapy as a background treatment. The choice of compression system will be guided by clinician and/or patient preference included in the centre’s formulary, such as Coban, Coban Lite, Coban Self-adherent, Comprilan, Lastodur, Profore, Profore Lite, Roselastic, Setopress, Surepress, compression hosiery and wraps. Any primary and secondary dressing may be used with the chosen compression system.
Throughout the trial, a participant’s general practitioner, nurse practitioner or specialist physician will be free to prescribe whatever concomitant treatments are necessary for the appropriate management of their patients. Concomitant medications, including complementary and alternative treatments, supplements and vitamins, will be recorded at each research visit.
Where a participant develops an infection related to the VLU during the course of treatment, the district nurses may use a medicated dressing for the treatment of the infection for recommended maximum period of 2 weeks. The medicated dressing will be selected from the formulary of medicated dressings available at each study centre, including silver, iodine, or honey-impregnated dressings as per clinician and/or participant preference. The participant will also be referred to a general practitioner or nurse practitioner for a prescription of an antibiotic where warranted. Each infection will be considered an adverse event, and participants and district nurses will be requested to notify the research nurse.
Data safety and monitoring
A DSMB has been established to review safety information. The DSMB consists of a senior biostatistician (Chair) and two other University of Auckland staff members with relevant expertise who are not involved with the trial and will meet not less than six monthly. The DSMB will draw up their own charter and will be free to review any information or study process in addition to reviewing safety data. The DSMB will make recommendations to the TSC on the continuation of the trial after each meeting or after a SUSAR.
The trial will be monitored by the project manager (AW) and the following information will be audited: admission to the district nursing service to verify the patient detail, record of consent, key baseline and outcome variables. Monitoring will begin once five participants have been randomised at a study centre.
Ethics and dissemination
Ethical approval for the trial was obtained on 6 January 2020 from the Northern B Health and Disability Ethics Committee (Reference 19/NTB/151) by the trial coordinating centre (National Institute for Health Innovation). The trial participants are covered by the government-funded national no-fault insurance scheme where participants suffer harm from their involvement in the trial. Locality approvals were obtained from each of the organisations responsible for the participating study centres prior to the trial start on 1 March 2020. Changes to the protocol are sought as required, incorporated into the trial registration as appropriate once notification of approval has been received, and communicated to the site investigators and research nurses. We will publish the peer-reviewed results of this trial in a journal within 1 year of completing follow-up. The results will be notified to the participants on trial completion.
Trial status
Recruitment to the trial commenced from 1 March 2020 and five participants were randomised by 19 March 2020. On 22 March 2020, due to the COVID-19 pandemic and evidence of community transmission in New Zealand, the New Zealand government declared ‘level 3’ restrictions (gatherings cancelled, public venues closed and deferment of elective healthcare procedures) with planned escalation to ‘level 4’ (level 3 restrictions plus suspension of all non-essential work, non-essential workers required to stay at home, closure of all educational institutions and reprioritisation of healthcare services, except for essential workers) within 48 hours. In anticipation of these restrictions, we decided to suspend recruitment into the Factorial4VLU trial on 20 March 2020. Recruitment restarted at the study centres on various dates in June 2020. The two Auckland centres had a further suspension of recruitment for 3 weeks in August–September 2020 due to a community outbreak and escalation of restrictions to level 3. Negotiations with the study centres have since resulted in agreement to continue participant recruitment up to and including during level 3 restrictions. Future participant recruitment will only be suspended if the government announces level 4 restrictions.
Acknowledgments
The authors wish to thank the site investigators (Sue Fenwick, Trish Johns, Natalie Scott, Cathy Hammond, Emil Schmidt) and the research nurses (Kate Wotton, Janet Patterson, Alice Bourke, Sian Ellis, Sue Lee), respectively, at Auckland District Health Board, Counties Manukau District Health Board, Capital and Coast District Health Board, Nurse Maude Association and Southern District Health Board for their continued support of the 4VLU research collaboration. The authors wish to acknowledge the contribution of the members of the 4VLU Data Safety Monitoring Board, Professor Thomas Lumley, Associate Professor Joanne Barnes and Dr Vanessa Selak.
References
Footnotes
Twitter @1CommonReader, @DrChrisBullen
Contributors AJ is the principal investigator, led all stages of the design development, grant application and protocol development. AJ drafted the manuscript. AW, CB, VP, JGMP, GL, JW, MK and JO’B all contributed to the study design, grant application and protocol development. All authors edited the draft manuscript and approved the manuscript for submission. AJ, AW, CB, VP, GL, and JW are members of the TSC. MK and JO’B are international advisors to the project.
Funding This trial is funded by a project grant (#19/069) from the Health Research Council of New Zealand (www.hrc.govt.nz). The hypochlorous acid is supplied by Te Arai BioFarma.
Disclaimer Neither the Health Research Council nor Te Arai BioFarma has any role in the conduct and analysis of the trial, nor will they have any role in the interpretation or decision to publish the findings from the trial.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note AJ is a registered nurse and professor at the School of Nursing as well as a senior research fellow at the National Institute for Health Innovation, both at the University of Auckland. AW is a senior project manager at the National Institute for Health Innovation at the University of Auckland. CB is a public health physician and director of the National Institute for Health Innovation at the University of Auckland. VP is a senior biostatistician at the National Institute for Health Innovation at the University of Auckland. JGMP is a registered physiotherapist and associate professor in the School of Nursing at the University of Auckland. GL is a medical oncologist, leader in Māori Health and qualified in health economics. JW is a geriatrician and executive officer of the HOPE Foundation for Research on Aging. MK is a reader in clinical physiology at Sheffield Hallam University. JO'B is an exercise scientist and registered nurse lecturing in nursing at the University of Tasmania.