Article Text

Abstract
Introduction Diabetic retinopathy (DR) is the main cause of adult visual impairment worldwide. Severe non-proliferative DR (sNPDR) is an important clinical intervention stage. Currently, panretinal photocoagulation (PRP) is the standard treatment for sNPDR. However, PRP alone cannot completely prevent NPDR progression. One explanation might be that PRP does not remove the detrimental vitreous that plays an important role in DR progression. Microinvasive pars plana vitrectomy (PPV) was shown to be a safe and effective method to treat late-stage proliferative DR (PDR) by completely removing the pathological vitreous. However, whether PPV is effective in controlling sNPDR remains unknown. In this trial, we aim to compare the effectiveness of microinvasive PPV with that of PRP for sNPDR progression control.
Methods and analysis This single centre, parallel group, randomised controlled trial aims to evaluate the clinical efficacy of microinvasive PPV in preventing the progression of sNPDR compared with PRP. A total of 272 adults diagnosed with sNPDR will be randomised 1:1 to the microinvasive PPV and PRP groups. The primary outcome is the disease progression rate, calculated as the rate of sNPDR progressed to PDR from baseline to 12 months after treatment. The secondary outcomes include the change in best-corrected visual acuity, re-treatment rate, diabetic macular oedema occurrence, change in central retinal thickness, change in the visual field, cataract occurrence and change in the quality of life.
Ethics and dissemination The Ethics Committee of Zhongshan Ophthalmic Center approved this study (2019KYPJ108). The results will be presented at scientific meetings and submitted for publication to peer-reviewed journals.
Trial registration number NCT04103671.
- diabetic retinopathy
- vetreoretinal
- ophthalmology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This would be the first prospective, randomised controlled trial to evaluate the effectiveness and safety of microinvasive pars plana vitrectomy, compared with panretinal photocoagulation, for the treatment of severe non-proliferative diabetic retinopathy (DR).
Early new vessels elsewhere are easily overlooked and may be difficult to distinguish from intraretinal microvascular abnormality based on the golden standard of DR grading, the Early Treatment Diabetic Retinopathy Study seven standard-field fundus photography.
The treatment of macular oedema (anti-vascular endothelial growth factor or steroids) can affect DR progression.
Since the participants would go to different hospitals and doctors to treat systemic diseases, this may cause certain biases, especially in the control of blood sugar.
The participants’ compliance with macular oedema treatment may be affected by economic factors, which may cause certain biases.
Introduction
Diabetic retinopathy (DR) is the most common microvascular complication of diabetes,1 2 and the leading cause of irreversible blindness in working-age adults worldwide.3 4 The prevalence of DR among adults with diabetes aged 45 years and older is 18.45% (13.16 million people) in China, and approximately 34.6% (93 million people) worldwide.5 According to the Early Treatment Diabetic Retinopathy Study (ETDRS) seven standard-field fundus photography and the international classification of DR, DR can be divided into two major stages—non-proliferative DR (NPDR) and proliferative DR (PDR).6 7 About 5%–10% DR develop vision-threatening complications, including PDR or diabetic macular oedema (DMO). Furthermore, data from the DR study (DRS) suggested that approximately 60% of the untreated diabetic eyes had shown progression after only 1-year follow-up.8 The ETDRS demonstrated that without timely treatment, the risk of progression to proliferative stage was high, with 45% of patients with very severe NPDR (sNPDR) and 15% of patients with sNPDR developing PDR within 1 year, and nearly half of the patients with PDR will experience profound vision loss.9
Panretinal photocoagulation (PRP) is the standard treatment for sNPDR and PDR.9 However, PRP reduces the risk of severe visual loss by only 50% and might cause some adverse effects, including peripheral visual field loss that precludes driving, nyctalopia, reduced retinal sensitivity and aggravated macular oedema that leads to vision impairment.10 Therefore, alternative treatments that would reduce the risk of progression of sNPDR to PDR, but with fewer adverse effects, are highly desirable.
The vitreous is shown to play a vital role in DR progression.11 Modifications in the metabolism and function of the retina during the progression of DR lead to changes in the structure and molecular composition of the vitreous. These changes cause pathological alterations to the diabetic retina, resulting in the activation of a vicious cycle that contributes to disease progression. In DR, the vitreous serves as a reservoir of pathological signalling molecules, such as vascular endothelial growth factor (VEGF) and many inflammatory factors that stimulate inflammatory and proliferative processes in the retina. These lead to macular oedema and neovascularisation.12 Many studies have demonstrated that hyperglycaemia cause crosslinking of collagen, and degradation of hyaluronic acid in the vitreous, thus contributing to diabetes-related synchesis, vitreoschisis and early posterior vitreous detachment (PVD).13 14 Furthermore, diabetes might cause adhesions in the posterior vitreoretinal region, leading to PVD, vitreous haemorrhage, epiretinal membrane formation and retinal neovascularisation.15 16 Therefore, early removal of pathological vitreous in DR can be expected to benefit the patients.
Pars plana vitrectomy (PPV) is widely used to treat vitreoretinal diseases by removing the pathological vitreous. It is used to treat vitreous haemorrhage, retinal detachment, eye trauma, PDR and even floaters.17 Vitrectomy represents an immediate and surgically induced PVD, and eyes with ischaemic retinal diseases, such as PDR, often demonstrate postoperative clinical stabilisation or improvement.18 Thanks to the advances in surgical equipment and technology, modern vitrectomy can achieve better vision recovery with smaller incisions and fewer complications. Microinvasive PPV was shown to be a safe and effective method to manage PDR by removing the pathological vitreous, proliferative membrane, VEGF and inflammatory factors.19
In summary, it is reasonable to expect that microinvasive PPV will be effective in controlling the progression of sNPDR by removing the pathological vitreous. However, whether microinvasive PPV is more effective than PRP remains unknown. Therefore, we plan to conduct this randomised controlled trial to compare the efficacy of PRP and microinvasive PPV in preventing sNPDR progression over 12 months.
Methods and analysis
Objectives
This study aims to investigate the efficacy of microinvasive PPV versus PRP in reducing the progression rate from sNPDR to PDR over 12 months. The secondary objectives are to assess the changes in best-corrected visual acuity (BCVA), visual field, central retinal thickness, quality of life, occurrence rates of cataract and DMO, and rates of re-treatment and adverse events (AEs).
Trial design
This trial is a single centre, parallel group randomised controlled trial. A total of 272 patients with sNPDR will be randomised 1:1 to the microinvasive PPV and PRP groups with 12-month follow-up after treatment. Interim analyses will not take place in this study. A completed Standard Protocol Items: Recommendations for Interventional Trials checklist for this trial is available (see online supplemental file 1).20
Supplemental material
Participant characteristics and setting
Patients will be assessed at the clinical research centre of Zhongshan Ophthalmic Center (ZOC) in Guangzhou, Guangdong, China, based on the inclusion and exclusion criteria.
Eligibility criteria
Inclusion criteria
Aged ≥18 years, men or women.
Diagnosed with type 1 or type 2 diabetes.
Presence of sNPDR according to the international 4-2-1 rule,7 with one or more of the following:
More than 20 intraretinal haemorrhages in each of the four quadrants.
Definite venous beading in two or more quadrants.
Prominent intraretinal microvascular abnormalities (IRMA) in one or more quadrants
ETDRS BCVA letter score >24 (Snellen equivalent 20/320) on the day of randomisation.
Clear media, good pupillary dilation and sufficient cooperation in administering PRP, and obtaining adequate optical coherence tomography (OCT) images and fundus photographs (FP).
Able and willing to sign informed consent.
Only one eye per participant will be included in this study. If both eyes are eligible, the selection of the study eye will be determined by the researcher. The other eye will be treated following the current guidelines.
PDR and sNPDR diagnosis will follow the Diabetic Retinopathy Preferred Practice Pattern 2019,21 according to the results of ETDRS seven standard-field FP.
Exclusion criteria
Unwilling or unable to provide consent for participation in the study, unwilling to submit to the randomisation process or unable to return for the scheduled protocol visits.
Participating in another clinical trial.
Has significant renal disease, defined as chronic renal failure requiring dialysis or kidney transplantation.
Has, in the investigator’s opinion, a condition that would preclude participation in the study, such as unstable glycaemic control. Poor glycaemic control is defined as intensive insulin treatment (a pump or multiple daily insulin injections) initiation within the last 4 months or a plan to do so in the next 4 months.
Has blood pressure >180/110 mm Hg. If blood pressure is maintained below 180/110 mm Hg with antihypertensive medication, the individual is eligible to participate.
Has a history of transient ischaemic attack, stroke, myocardial infarction, acute congestive heart failure or other acute cardiac event requiring hospitalisation within the past 4 months.
Received systemic pro-VEGF or anti-VEGF treatment within the past 4 months.
For women of childbearing age: potential study participants would be questioned about pregnant, lactating or intending to become pregnant within the next 3 years. The investigator would determine when a pregnancy test is required.
Underwent PRP with laser photocoagulation in over 100 points.
Has macular oedema caused by reasons other than diabetes. This could be when the primary cause of macular oedema is ocular surgery (eg, cataract extraction) or presence of vitreoretinal interface abnormalities or disease (eg, epiretinal membrane or vitreous macular traction) as suggested by OCT.
Has another coexisting ocular disease that, in the investigator’s opinion, might decrease visual acuity during the study. These include retinal artery or vein occlusion, age-related macular degeneration, uveitis and neovascular glaucoma.
Has substantial cataract that, according to the investigator’s judgement, is likely to decrease visual acuity by more than three lines (ie, the cataract reduces the visual acuity to 20/40 or worse).
Has a history of corticosteroid treatment (peribulbar or intravitreal) in the past 4 months.
Received intravitreal anti-VEGF treatment in the past 2 months.
Had a major ocular surgery (eg, vitrectomy, scleral buckling, cataract extraction or any intraocular surgery) in the past 4 months or plan to have one within the next 6 months.
Underwent yttrium aluminum garnet capsulotomy within the past 2 months.
Aphakia present.
Has, in the investigator’s opinion, uncontrolled glaucoma.
Has severe external ocular infection, including conjunctivitis and substantial blepharitis.
Recruitment procedure
Potentially eligible participants will be screened and assessed at the ZOC after written informed consent is obtained (seeonline supplemental file 2). Patients who meet the eligible criteria will be enrolled in the randomised trial. The participant flow diagram is shown in figure 1.
Supplemental material
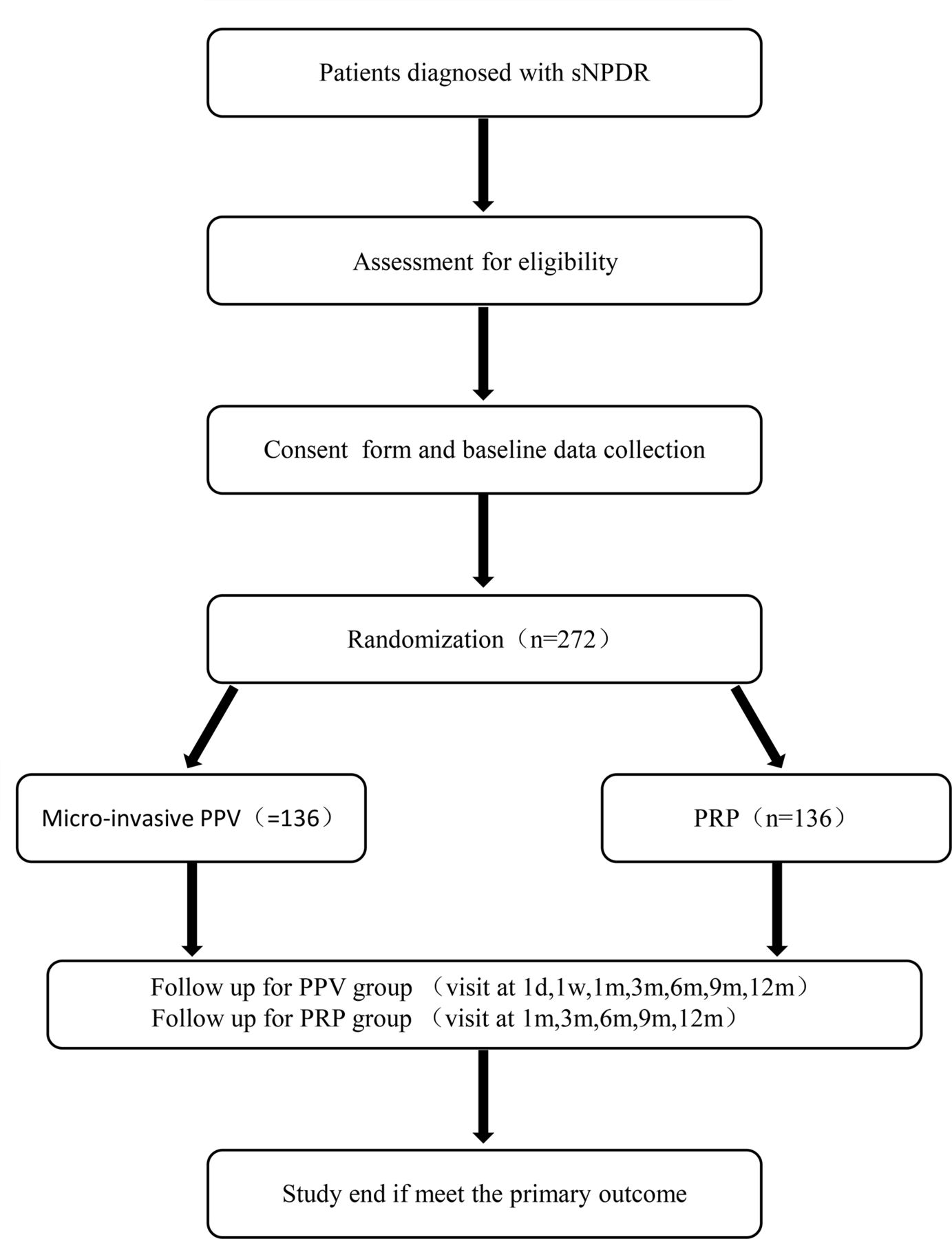
Participant flow diagram. d, day; m, month; PPV, pars plana vitrectomy; PRP, panretinal photocoagulation; sNPDR, severe non-proliferative diabetic retinopathy; w, week.
Participant timeline
The schedule of enrolment, interventions, assessments and visits for participants is shown in table 1.22
Schedule of enrolment, interventions and assessments
Sample size
The sample size estimate has been computed for the primary study objective, to determine whether disease progression rate in the PPV group is superior to that in the PRP group at 1 year. The sample size calculations are based on the published Diabetic Retinopathy Clinical Research Network clinical trials.9 23 Disease progression is defined as the transition from sNPDR to PDR according to the international definition of DR.6 It is assumed that the 1 year cumulative sNPDR progression rate after the PRP treatment would be approximately 40%, and that after the PPV treatment would be 20%. By using a balanced design, 109 participants per study group will be required to achieve 90% statistical power at a two-sided significance level of 0.05. Thus, allowing for 20% of participants loss to 1-year follow-up, 272 participants are required for this study.
Randomisation
The study statistician will generate a random sequence using an online random number generator (www.randomization.com), with A and B representing the groups. After informed consent is obtained, eligible participants will be randomised 1:1 with block sizes of four and eight to the microinvasive PPV or PRP groups. A nearly equal number of participants will be assigned in both groups, and the investigator will not be able to predict the next participant’s assignment. The patient and investigator will be informed about the allocated group after the randomisation.
Blinding
This is an open-label trial. Participants and physicians are not masked to the treatment assignment. The primary study outcome is based on seven standard-field FP, which will be read by masked graders. Masked ophthalmic technicians will obtain other important variables, such as OCT, BCVA, intraocular pressure (IOP), visual field and questionnaire scores following the standard operating procedures (SOP). Finally, the study data will be analysed by masked statisticians.
Collection of biological specimens
Collection of biological samples (vitreous fluid and blood) will be based on the patient’s wish and signed informed consent. Each patient will be able to voluntarily choose whether to take blood samples during the baseline examination. Vitreous fluid samples will be collected during the microinvasive PPV procedures. The biological samples will be stored at −80°C in a biological sample bank pending analysis.
Investigations
Six visits over the 1-year follow-up period are planned. All the examinations will be conducted by experienced technicians trained according to the study SOP. These examinations will include:
Seven standard-field FP
The images will be obtained using a digital fundus camera (Zeiss VISUCAM 224, Carl Zeiss Meditec, Germany) following the ETDRS study operating specifications,9 as shown in figure 2. Briefly, compound tropicamide eye drops will be instilled into the eye at least three times before the procedure, every 10 min, to attain satisfactory pupil dilation. The camera software will merge the seven standard-field fundus images. The criteria for acceptable images are correct orientation and clear retinal visualisation. The reading specialists will determine the result independently, following the international DR classification criteria. The FP reading panel will consist of three experienced vitreoretinal specialists. Two specialists will read every FP image. If the results are inconsistent, the third will determine the final result.

{kind=link}
{kind=link}
Seven standard-field fundus photography. (A) Pattern diagram, field 1 is centred on the optic disc, field 2 on the macula, field 3 is temporal to the macula and fields 4–7 are tangential to horizontal lines passing through the upper and lower poles of the disc and to a vertical line passing through its centre. (B) Stitched fundus photograph.
OCT
Images will be acquired through the dilated pupil with a spectral-domain OCT device (Cirrus SD-OCT 5000, Carl Zeiss, Germany). The technicians will verify the OCT examination accuracy by ensuring that it is centred and of adequate quality.
Visual field testing
All tests will be performed by Humphrey Perimeters (Carl Zeiss Meditec, Dublin, California, USA) with 30-2 and 60-4 fast patterns. Unreliable tests will be repeated until obtaining a reliable one with false-positive and false-negative errors less than 15%, and fixation loss less than 20%. No more than five attempts to achieve the two (at least) reliable fields are allowed.
Fundus fluorescein angiography
All subjects will go through fundus fluorescein angiography examination (Spectralis HRA+OCT, Heidelberg, Germany) unless they are allergic to fluorescein sodium.
Visual acuity
BCVA will be tested at 4 m, using the ETDRS LogMAR chart (Precision Vision, Villa Prak, Illinois, USA), after performing optometric assessment and refractive error correction.
Intraocular pressure
Non-contact computerised tonometer (CT-1, Topcon, Japan) will be used to measure the IOP. All subjects will be measured at least three times until a difference of less than 5 mm Hg is reached. The average value of three consecutive measurements will be recorded.
Slit-lamp biomicroscopy
A thorough examination of the eyes will be operated using a slit-lamp (BQ-900, Haag-Streit, Switzerland) and a 90D indirect ophthalmoscopy lens.
Questionnaires
Translated Chinese version of the National Eye Institute Visual Function Questionnaire-25 and the registered EuroQol-5 Dimensions five-level will be administered according to the study schedule.24 25
Measurement of blood pressure
An electronic sphygmomanometer will be used to measure blood pressure.
Biochemical tests
Haemoglobin A1c, blood creatinine, urea nitrogen, triglycerides and cholesterol will be tested.
The classification of DR
The grading and classification of DR will follow the International Clinical Diabetic Retinopathy and Diabetic Macular Oedema Disease Severity Scales.7
Intervention description
Microinvasive PPV
Standard 25G PPV will be performed by an experienced surgeon under local or general anaesthesia. After clearing the central vitreous, a complete PVD will be achieved with aspiration to remove the tightly attached posterior hyaloid. The vitreous will be removed by a high-speed vitrectomy surgical system (Constellation Vision System, Alcon Laboratories, Fort Worth, Texas, USA). The eye will receive no laser treatment during the surgery and the vitreous cavity will be filled with perfusion fluid at the end of the procedure. The surgery will be performed within 4 weeks after randomisation.
Pan-retinal photocoagulation
A VISULAS 532s diode laser device (Carl Zeiss Meditec) will be used in PRP treatment. Compound tropicamide eye drops will be instilled to attain satisfactory pupil dilation, and topical anaesthesia will be administered prior to the procedure. A total of 1200–1600 burns with a spot size of approximately 500 µm will be applied on the retina over 50–200 ms. The power will be modified arbitrarily until an effective retinal laser spot is obtained. The patient will receive the first laser treatment within 3 days of randomisation. The standard PRP treatment will be completed in three sessions within 4 weeks.
Criteria for discontinuing or modifying allocated interventions
The emergence of PDR is the endpoint of this trial. Participants with PDR will withdraw from the study and undergo further treatment according to current guidelines. Patients with other illnesses that would prevent attending follow-up visits or death will also withdraw from the study. DMO, including newly occurring and exacerbation of existing DMO, will be evaluated and treated according to current guidelines 3 months after completing the intervention. Newly occurring DMO is defined as OCT central retinal subfield thickness (CST) >250 µm on Zeiss Stratus OCT (or men >305 µm and women >290 µm on Zeiss Cirrus SD-OCT) and BCVA<78 letter score (approximate Snellen equivalent 20/32), exclusion of other macular oedema causes and absence of the DMO at baseline. Exacerbation of DMO is defined as DMO that was present at baseline (CST >250 µm and BCVA <78 letter score), and DMO-induced continued deterioration in CST or BCVA 3 months after the intervention, as determined by the investigator. Deterioration is defined as an increase in CST by more than 50 μm compared with baseline, or more than five letters of visual loss in two consecutive follow-up visits.
Strategies to improve adherence to interventions
The trial is implemented under the supervision of the Ethics Committee of ZOC. The research team will submit yearly research progress reports and accept random checks at any time.
Relevant concomitant care permitted or prohibited during the trial
The patient’s blood glucose control will follow the treating physician’s recommendations. The patients will inform the research doctors before using any medication. Systemic steroids or pro-VEGF drugs are prohibited during this trial.
Provisions for post-trial care
At the end of this trial, participants will be able to choose to enter a 2-year extended follow-up period. For those unwilling to continue, follow-up observations and further treatments will be performed following the current guidelines.
Primary outcome measures
The disease progression rate will be determined by the number of patients in each group in whom the disease progresses, following the specific treatment, during the 1-year follow-up period. DR progression is defined as the presence of one or both the following:
Neovascularisation.
Vitreous/preretinal haemorrhage.
Secondary outcome measures
All secondary outcomes compare the groups for change from baseline to the assessment after 12 months or the occurrence over these 12 months.
Change in BCVA in each group.
Re-treatment occurrence, including supplemental PRP or vitrectomy.
DMO occurrence in each group.
Central retinal thickness change.
Visual field change.
Cataract occurrence.
The change in the quality of life, as assessed by the questionnaires.
Safety evaluation
An evaluation of the safety of the procedures, including any intervention-related complications, progression of the disease itself will be conducted and will cover:
Corneal injury: corneal injury will be sought via slit-lamp microscope examination.
Ocular hypertension: IOP will be measured by tonometer.
Lens injury: lens injury will be determined following slit-lamp examination.
Macular oedema: an OCT scan will be applied to evaluate the status of the macula.
Retinal injury: a fundus examination will be conducted to evaluate whether retinal injury has occurred.
Adverse events
Complications, such as death, blindness, endophthalmitis, retinal detachment, vitreous haemorrhage and choroid detachment are serious AEs that need to be reported to the Ethics Committee of ZOC within 24 hours. If AEs occur, the safety of the participants would be the first concern, and any emergency treatment will be referred to an ophthalmologist or physician.
Study governance
An independent Trial Steering Committee and Data Monitoring Committee, consisting of ophthalmologists, statisticians and epidemiologists, will oversee the study integrity, recruitment, randomisation, interventions and analysis. The coordinating centre, consisting of the research team (assistants, nurses and doctors) and logistic support staff will be responsible for coordinating arrangements during this trial. The research team members will attend weekly meetings to discuss problems encountered during implementation and improvement measures. Any trial protocol changes need to be submitted to the ethics committee for review and approval before implementation. All protocol violations during the trial will need to be documented and explained. Violations of the protocol should be reported to the ethics committee. All serious AEs should be reported to the ethics committee within 24 hours. The study may be prematurely discontinued by the ethics committee, or for reasons reported to the Trial Steering Committee.
Data collection and monitoring
Trained clinical research associates at the clinical research centre of ZOC will collect and examine the data following the trial schedule. All data will be entered into an electronic database. The electronic materials will be backed up and paper materials of the participants will also be preserved. The data manager will continuously monitor the data to control its quality. During or at the end of the trial, collected data suitable for analysis will be anonymised before use. Access to the final data set will be limited to the trial administrator and the statistician. The Data Monitoring Committee will supervise the entire data collection process.
Data analysis
Baseline demographic and clinical characteristics will be presented as frequency (percentages) for categorical variables, and means (SD) or median (IQR) for continuous variables. No statistical tests will be performed for the differences between the study groups on any of the baseline variables.
The primary and secondary outcome analyses will be conducted according to intention-to-treatTT) criteria. All missing data will be imputed by last observation carried forward or multiple imputations. The multiple imputation approach creates 20 copies of the data, in which missing values are imputed by chained equations. The final result is obtained by averaging these 20 data sets using Rubin’s rules.
The unadjusted difference and 95% CI between the outcomes in the study groups will be calculated using the two-proportion z-test for the primary outcome and binary for categorical or two-sample t-test for continuous secondary outcomes. Histograms and the Shapiro-Wilk test will be used to check the t-test normality assumption. If the normal distribution assumption is not met, a non-parametric analysis will be considered. Univariate and multivariate logistic regression analyses will be performed for the primary outcome. The study group and all significant variables with p<0.20 in the univariate regression analyses will be included in the multivariate regression analysis. The OR and 95% CI will be estimated, adjusting for baseline measures, where appropriate.
AEs will be compared between the groups by χ2 test or Fisher’s exact test. All AEs related to the study and the clinical examination reporting abnormalities will be described in a table.
All statistical analyses will be performed at a two-sided significance level of 0.05, using a commercially available software package (Stata 16, StataCorp, College Station Texas, USA).
Ethics and dissemination
The Ethics Committee of ZOC approved this study and the consent form and study protocol were also approved by the local institutional review board (see online supplemental files 3 and 4) (2019KYPJ108). The chief investigator will ensure that this study is conducted in accordance with the principles of the World Medical Association Declaration of Helsinki. Written informed consent will be obtained from eligible participate. A copy of the signed consent will be given to the patients and a second copy will be stored in the study master file.
Supplemental material
Supplemental material
Dissemination
The study results will be presented at scientific meetings and submitted for publication in peer-reviewed journals.
Discussion
PRP is currently the standard treatment for sNPDR and early PDR. However, PRP is not enough to prevent DR progression. In our clinical observation, more than 50% of the patients progress to PDR within 8 years of the PRP treatment, requiring vitrectomy (unpublished).
Previous studies have shown that vitreous lesions play an important role in DR progression and prognosis.11 PVD increases vitreous cavity oxygen content and reduces VEGF concentration, which might be beneficial for retinal ischaemic diseases. As vitrectomy is the most direct form of PVD, it is reasonable to expect it to control DR progression by removing the pathological vitreous, decreasing VEGF concentration in the vitreous cavity, improving the retinal feeding state and reducing neovascularisation.
A randomised controlled trial on the optimal sNPDR treatment is required, as such is currently lacking. This study would be the first prospective, randomised controlled trial to compare the effectiveness and safety of microinvasive PPV and PRP in sNPDR treatment. The findings are expected to provide evidence that microinvasive PPV is a better treatment option for sNPDR.
Patient and public involvement
The patient and the public were not involved in the design and will not be involved in the study conduct. We will promote the knowledge of DR to patients with diabetes and the public through online and offline lecture presentations. Besides, our research results will be disseminated to peers through academic conferences and scientific publications. We will also share information with study participants and patients.
Trial status
It is ongoing. The current protocol is Version 1.0, 20190724. Recruitment for the study began in October 2019. The first inclusion was on 3 December 2019. Due to the outbreak of the 2019 new coronavirus, the enrolment is relatively slow currently. As the epidemic is brought under control, there will be more subjects participating in this trial. It is anticipated that the study will reach the recruitment target of 272 participants by September 2021.
Acknowledgments
Special thanks to Professor Xiaoyan Ding for her great help in this trial. And also, we would like to thank all research assistants and nursing staffs involved in this trial, who contributed to the practical organisation and execution of this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
WZ and SC are joint first authors.
Contributors Tao Li, Yizhi Liu put forward the conception of the study. Tao Li, Yizhi Liu, Ying Lin, Bingqian Liu, Xiaohu Ding, Kunbei Lai, Sainan Xiao, Jizhu Li, Yuqing Wu, Yuan Ma, Lin Lu, Wenbin Zheng, Shida Chen together designed the study, made the study protocol and recruit the patients. Tao Li, Wenbin Zheng, Shida Chen wrote the manuscript. Lin Jin performed all the statistic related design. All authors read and approved the final manuscript.
Funding This study was supported by grants from the Guangdong Province High-level Hospital Construction ProgramProgramme (303020103) and the National Natural Science Foundation of China (81670872). Neither the sponsor nor the funder had any role on the study design, collection, management, analysis of data and the writing of this manuscript.
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer-reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.