Article Text

Abstract
Introduction The power of ‘real world’ data to improve our understanding of the clinical aspects of multiple sclerosis (MS) is starting to be realised. Disease modifying therapy (DMT) use across the UK is driven by national prescribing guidelines. As such, the UK provides an ideal country in which to gather MS outcomes data. A rigorously conducted observational study with a focus on pharmacovigilance has the potential to provide important data to inform clinicians and patients while testing the reliability of estimates from pivotal trials when applied to patients in the UK.
Methods and analysis The primary aim of this study is to characterise the incidence and compare the risk of serious adverse events in people with MS treated with DMTs. The OPTIMISE:MS database enables electronic data capture and secure data transfer. Selected clinical data, clinical histories and patient-reported outcomes are collected in a harmonised fashion across sites at the time of routine clinical visits. The first patient was recruited to the study on 24 May 2019. As of January 2021, 1615 individuals have baseline data recorded; follow-up data are being captured and will be reported in due course.
Ethics and dissemination This study has ethical permission (London City and East; Ref 19/LO/0064). Potential concerns around data storage and sharing are mitigated by the separation of identifiable data from all other clinical data, and limiting access to any identifiable data. The results of this study will be disseminated via publication. Participants provide consent for anonymised data to be shared for further research use, further enhancing the value of the study.
- multiple sclerosis
- adverse events
- clinical pharmacology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
All patients with multiple sclerosis eligible for disease modifying therapy under the care of participating centres can take part in this study.
Selected clinical data recorded during episodes of routine care are uploaded to a study specific database, allowing an assessment of data completeness and quality.
Potential concerns around data storage and sharing are mitigated by the separation of identifiable data from all other clinical data, and limiting access to any identifiable data.
Data are representative of patient outcomes in specialist MS centres, where an increasing proportion of MS care is provided; patients at non-specialist centres are not enrolled, so their outcomes will not be captured.
A strength of the study recruitment processes is the consent for contact regarding additional studies; the study database thus provides a rich resource for future research.
Introduction
Real world data in multiple sclerosis
The power of ‘real world’ data to improve our understanding of the clinical aspects of multiple sclerosis (MS), including the relative risks and benefits of disease modifying treatments and efficacy in a UK population with MS, and the future impact of these data on the care of people with MS is starting to be realised. However, the utility of large datasets is significantly influenced by the methods used to capture date, as well as the overall validity of these data. Concerns regarding data quality and international applicability have limited the translation of findings from large datasets to clinical practice across medicine. A huge number of unanswered questions remain around risk stratification, treatment sequencing, and outcome monitoring, which cannot be answered using a traditional randomised control trial (RCT) design. Furthermore, long-term and/or rare outcomes are almost impossible to detect via RCTs. Historically, phase 4 observational studies have been used to study such endpoints. However, these also have limitations due to high drop-out rates and an inherent lack of power to detect rare outcomes, or to evaluate risks associated with prior or subsequent treatment decisions, or comorbid medical conditions.
‘Real world data’ refers to data gathered in the course of an individual patient’s routine clinical care. The aim is not to discriminate based on age, comorbid conditions or other such factors that may influence trial eligibility.1 These data are by nature highly confounded—which brings significant statistical concerns—but also enables observations relevant to clinical practice. Such data enable the study of specific areas that cannot be addressed via traditional routes.
A number of large real world MS-specific data initiatives already exist. National registries from European countries, including Sweden, France and Italy have significantly improved our understanding of MS disease trajectories and the influence of treatment on these.2 MSBase is a collaborative, international disease registry, which captures a range of MS-related data from a large number of countries.3 More recently, processes for formal data sharing have started to enable the pooling of data from five large registries (Sweden, France, Italy, Denmark, MSBase).4
Across the UK, harmonised data gathering has been limited to date. While many databases exist, most lack the granularity needed for the objectives of this study. For example, the database for the Blueteq web-based prior approval system identifies people who are prescribed disease modifying therapy (DMT), but does not capture safety data. Rigorous data collection and prospective data entry is time consuming, and frequently does not provide short, or even medium, term benefit to clinicians. Linking data entry to DMT funding rebates brings a clear risk of biasing data. While many of the concerns regarding missing or incomplete data can be overcome with large numbers of participants,5 in order to overcome reporter bias when examining potentially rare or serious events, data collection and verification needs to be rigorous.
Given the relatively homogeneous practices in DMT prescription across the UK, driven by national prescribing guidelines,6 the UK provides an ideal arena from which to gather MS outcome data.
Need for pharmacovigilance in MS
The therapeutic armoury in MS has increased rapidly, but real-world data concerning safety is limited. The first treatments to be introduced (interferon-beta preparations and glatiramer acetate (Copaxone)) have well-established safety records.7 Progressive multifocal leukoencephalopathy risk limits the long-term use of natalizumab in many patients.8 There are safety signals from clinical trials and early clinical experience suggesting both lymphopaenia and increased infection rates with newer MS therapies.7–12 The risks of serious adverse events (SAEs) with individual treatments over a relatively short duration have been reported in clinical trials (table 1). The rates at which these occur in the real-life treated UK population which includes a population with a wide range of age and comorbidities, is as yet, unknown. The current adverse event reporting system (yellow card system12) has a high risk of missing longer-term rare adverse events; as such there is an urgent need for a formalised reporting system. The yellow card system is voluntary, and medicines that are licensed for less than 2 years are highlighted. Awareness raising has a substantial impact on both the quantity and quality of reports13; highlighting under-reporting and bias in reports.
Rates of SAEs in treatment and control groups documented in published clinical trial data
As the number of patients taking newer MS treatments increases, so does our clinical experience. Alongside this, concern regarding longer-term effects of treatments and the cumulative risk associated with treatment switching is increasing (personal communication, MHRA). Counselling patients regarding switching DMT typically uses information available from (often recent) clinical experience, case reports and occasional small case series.
In a pharmacovigilance study, patients on established first-line treatments provide a useful reference cohort, as the safety and effectiveness of these medicines are well understood. They form a readily characterised group; patients tend not to be de-escalated to first-line injectable therapies outside of specific situations, such as when planning pregnancy or with identification of an active malignancy (observations from authors’ clinical experiences). While there are obvious limitations to indexing outcomes associated with newer treatments to these reference populations (eg, disease and demographic characteristics may be different, as patients in the reference groups are likely to have lower baseline disease activity than those receiving highly active therapies), they provide a pragmatic approach for an observational real-world study.
Existing models of disease-specific studies evaluating treatment-associated events
National pharmacovigilance is not a novel concept and can contribute to improved safety for patients. Differences in susceptibilities and pharmacological responses, diagnostic and prescribing practices, lifestyle and patterns of medicine adherence all can vary between countries. Both observational registries and prospective cohort studies are well-established in rheumatology and dermatology, where they may be linked to medication funding, and these provide a wealth of data regarding the safety profile of biological agents.14–16
National, automated data capture systems, some of which are disease specific, are increasing. NHS Digital now curates a number of national data collections.17 However, MS outcomes are poorly coded in primary care datasets, and individuals with MS who develop cancer and are entered onto cancer registries do not have detailed data regarding prior DMT use recorded. It is thus not possible to use existing registries to study clinical endpoints in MS in detail; however, these registries provide a resource to mitigate against loss to follow-up via data linkage.
Methods and analysis
Rationale and study design
The OPTIMISE:MS study is a pragmatic, prospective observational cohort study (19/LO/0064) planned to run for 7 years in the first instance, with the potential for longer follow-up subject to funding. It aims to estimate the frequency of SAEs with DMT use in routine clinical practice in the UK. All individuals potentially eligible for MS DMT are eligible for this observational study, regardless of any decision regarding DMT use (figure 1). OPTIMISE:MS will recruit people with MS from major MS care clinics across the country.
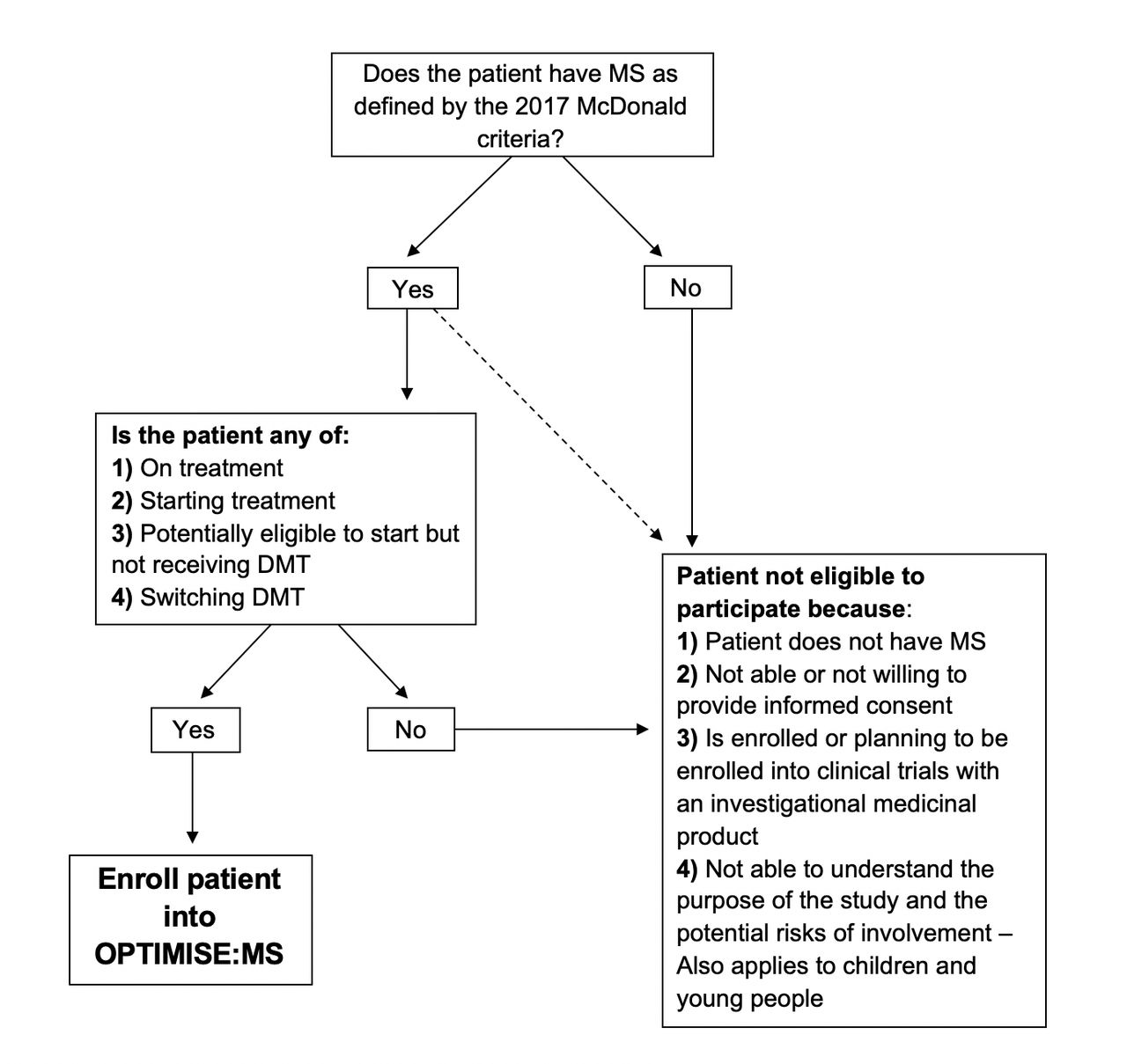
{kind=link}
Participant entry criteria flowchart. DMT, disease modifying therapy; MS, multiple sclerosis.
All participating sites have a site initiation visit prior to approaching any potential participants. Sites receive training in the study aims, and how these will be met using the databases. Database training is provided, with ongoing support from the central team for both technical issues and support for clinical coding or event adjudication. Participating sites are reimbursed for their participation in the study based on numbers recruited and followed up. Set up costs are covered at the time of site initiation, followed by per-participant recruitment costs and subsequent follow-up costs. These have been calculated according to the anticipated time for data capture and entry. Patients are not reimbursed for participation in the study, as other than the time taken to consider the study and provide consent, no additional demands are made of them.
The primary aim of this study is to characterise the incidence and compare the risk of SAEs in people with MS treated with DMTs (comparators will be an untreated cohort, and a cohort treated with first line injectable DMT). An SAE in this context is an adverse event resulting in death, requiring inpatient treatment or prolongation of existing inpatient treatment or that results in persistent or significant disability or incapacity. SAEs of particular interest in this study are any infections requiring hospitalisation, any opportunistic infection (ie, typically associated with immunosuppression), any other SAE believed to be related to treatment (eg, lymphoma, non-melanoma skin cancer), any MS relapse and death. Additionally, rates of symptomatic COVID-19, outcomes associated with SARS-CoV-2 infection and relationship to DMT use and/or immunosuppression will be captured. There has been the additional potential to capture data regarding the frequency of asymptomatic COVID-19 infections and responses to vaccination alongside this.
Selected clinical data, clinical histories and patient-reported outcomes will be collected in a harmonised fashion across sites at the time of routine clinical visits or in direct follow-up (for laboratory or paraclinical testing). SAEs will be classified according to MedDRA. No additional clinical visits are mandated as part of this pragmatic study. Based on usual (recommended) clinical practice in the UK, we anticipate follow-up clinical visits for patients enrolled in this study at least on an annual basis. Current National Institute of Health and Care Excellence criteria for the management of MS in the UK along with NHS England DMT financial rebate systems (Bluteq) require at least annual review of all patients with MS. The requirement for at least annual review has thus been set in line with minimum standard clinical practice.
MRI scans and laboratory tests recommended for treatment monitoring will be performed by clinical centres at intervals based on their usual clinical practice and clinical indications in the patients. While sites can enter MRI data into the database, there is no mandate for MRI monitoring within the study protocol. Sites are provided with financial support to complete these follow-up data in line with the compensation for completing DVLA forms. As part of site support and data queries, the return rate for follow-up data and completeness of these data will be audited by the central study team. As all UK patients have a permanent NHS number allocated at birth or migration, participants who move around the country can continue to be monitored.
Concerns regarding loss to long-term follow-up can be mitigated to some degree via data linkage with national systems. Following appropriate consent, an individual’s NHS number can be used to link individuals via digital.NHS, Hospital Episode Statistics and the UK Cancer Registry, potentially using HDR-UK infrastructure, to support follow-up of SAEs.
This study also has the potential for patients to report on the impact of the disease on their daily life via patient-reported outcome measures. However, these would require a separate, patient focused database portal along with additional consent. This remains an aim for the database. However, given COVID-19-related delays to initial recruitment and pressure on sites, the current focus is on collecting accurate and complete physician-reported data.
Data flow and security
The OPTIMISE:MS database (online demo version available via https://www.optimise-ms.org/demo/) has been designed to enable electronic data capture and secure data transfer. Data are entered onto site-specific laptops at individual sites, and transferred securely on a regular basis. Every data entry is time stamped according to both date of data entry and date of data collection, to allow for audit and identification of retrospective data entry. Personally identifiable information including NHS number is accessible only to the clinical care team at each site and to those members of the central analysis group responsible for follow-up of SAEs. Where insufficient data are entered on a participant with an SAE, this will be followed up by the central analysis team as part of quarterly data quality review.
The majority of participant data are stored in a secondary database, in which each subject is pseudonymised and the NHS number and name are not available. This database will be used for analyses performed by the OPTIMISE analytical group and collaborators. A participant data deletion mechanism is incorporated into each form of the database. Data for any subject who withdraws consent and whose data are deleted from the database held at any participating site also will be deleted automatically from the two forms of the database held at the central analysis site.
Patient and public involvement
Patients were involved early in the conception of this study. The study was discussed with patient representatives, who gave feedback around the kind of data that they would be willing to share, and the desired frequency of contact. The main concern of the patients that we consulted was that their data were to be used for the benefit of people with MS, and not for profit or other third party gain.
OPTIMISE database and core dataset
While software tools for MS patient data entry and curation have been generated before, none are in widespread use. There is no single electronic patient record (EPR) in use across the UK NHS, and using individual EPR systems to collect MS-specific data is likely to be both unacceptably time consuming and costly. One major challenge of bespoke MS data collection systems is the need to balance comprehensiveness of data collection with the sustainability of data entry in a busy clinic environment. A further challenge is to ensure compliance with basic requirements for data security and auditing and the ability to remove data if consent is withdrawn.
The OPTIMISE database is used to capture both minimum baseline characteristics and ongoing and follow-up data. These include current treatment category, gender, ethnicity, dominant hand, country of origin, primary MS diagnosis, time since diagnosis, age, physical and performance measuresExpanded disability status scale (EDSS), relapses within the past 2 years, SAEs, malignancies, opportunistic infections, liver function, brain MRI, total white cell and lymphocyte count (linked to individual level immunosuppression) and anti-JCV antibody status (table 2).
Core baseline characteristic and ongoing / follow data capture for OPTIMISE:MS patient cohort
Participating sites are expected to upload all of the clinical and paraclinical data collected to the central analysis site on a quarterly basis. Quarterly data checks allow sites to focus on ensuring their data are of high quality, and reduces the number of data queries raised to sites. Visit frequency for patients was determined by usual clinical practice and completeness of data and other quality control assessments are performed on a quarterly basis by the central study team located at Imperial College London. Where data are incomplete in a significant number of records in a single upload, the site is contacted for remedial action to be taken. A strength of this study is that data queries regarding missing or implausible data are sent directly to sites, allowing entries to be reconfirmed as required. All changes made in response to these are timestamped and recorded in the database, so that sites with high levels of implausible or missing data can be identified for additional training and support.
Statistical analysis
The statistical analysis will focus on the detection of adverse event signals by identifying specific DMTs and DMT sequences associated with disproportionate numbers of events (relative to the overall study population). With 4000 patients followed up for 5–7 years, this study will generate between 20 000 and 28 000 patient years of follow-up. Site prescribing practices and geographical location will be considered in the analysis, as prescribing is not uniform across the UK, and location may impact on infection risk (rural vs urban).
A number of disproportionality measures will be applied in parallel to increase sensitivity, including the reporting OR, empirical Bayes geometric mean and information component18 (all originally designed for spontaneous report data, but here adapted to the longitudinal data collected within OPTIMISE:MS), and the incidence rate ratio. Signals will be triggered if the lower one-sided 95% confidence limit for any disproportionality measure exceeds the null value of no association. Two methods will be used to screen out false positive signals. First, a minimum number of events will be imposed in order to eliminate false signals due to random noise when event counts are low. Second, a chronological filter based on the LEOPARD methodology will be applied.19 The LEOPARD filter assesses whether an adverse event occurs more often before or after the prescription of the treatment with which it appears to be associated. This can help to avoid protopathic bias, in which a causal relationship is falsely inferred between an adverse event and the commencement of treatment when in fact both were caused by the underlying progress of disease or other factors.
Poisson (or negative binomial) regression and survival analyses will be carried out to evaluate rates of adverse events, relapse, disability progression, new MRI lesions, mortality, lymphopaenia and liver enzyme elevation in subjects receiving any newer DMT in comparison to those receiving first-line injectable DMTs or no treatment. These analyses will incorporate subject-level covariates to adjust for demographics and disease history.
Secondary analyses will use additional models to explore the relationships between DMT use and outcomes, including whether effects of DMTs persist after treatment cessation/switch. Several exposure definitions will be applied in parallel. Current exposure models will use the subjects current treatment category at the time of event. Recent exposure models will be dependent on both the current treatment category and, if applicable, the previous treatment category within the preceding 6 months. Cumulative exposure models will include the total cumulative exposure to each treatment class in patient-months, and time-weighted cumulative exposure models will weight according to both time on treatment and time from exposure to event, with more weight assigned to recent exposures than historic. In these analyses subjects will not be censored at treatment cessation or switch, so each subject can contribute multiple periods of follow-up with exposure to different treatment categories.
Entry criteria and follow-up data
All patients with MS as defined by the 2017 McDonald criteria and who are eligible to receive DMT reimbursement from NHS England are eligible for entry into the observational study. This includes patients who are on treatment, as well as those starting, potentially eligible to start but not receiving DMT, or switching DMT. Patients with either relapsing-remitting or progressive MS who meet these criteria are eligible for inclusion. This allows the inclusion of new groups of patients as new DMTs become available. This study will also enrol children aged over 11, although children and adolescents who are enrolled should be treated according to an established protocol within the NHS.
Patients who are not able or not willing to provide informed consent will be excluded from the study. Participants enrolled or planning to be enrolled into a clinical trial of an investigational medical product will be excluded for the duration of their participation in the trial (including any extension study).
Enrolling patients via participating major MS centres means that those patients who are treated only general neurology clinics, and/or are not seen in these centres are not included in this study. Centres were selected according to their caseload, with centres able to meet a recruitment target of at least 300 patients selected to take part in the study. Given the infrastructure support required for consent and ongoing data entry, a minimum number of recruits was needed to make the study viable within each centre. An advantage of this approach is that all of the major MS centres selected treat patients with the full range of MS DMTs. However, this approach also introduces a potential source of bias. Patients not seen at MS centres are more likely to be on either no treatment or first line injectable treatments, whereas those treated at MS centres potentially have access to highly active treatment earlier in their disease course. However, early data (table 3) suggest that this strategy has been successful in recruitment patients across a range of DMT.
Key baseline attributes of the 1615 subjects recruited as at January 2021
Recruiting during the COVID-19 global pandemic
Due to the COVID-19 global pandemic and two subsequent lockdowns within the UK, telephone and video consultations became standard for outpatient care, with face to face appointments only taking place sporadically. Redeployment of research staff to COVID-19 studies further impacted on recruitment. The lack of face-to-face contact has prevented direct assessments of EDSS.
Study amendments have been made and approved to accommodate remote enrolment of patients using postal consenting methods. The use of remotely collected EDSS20 has facilitated monitoring of patients at a number of sites.
Subjects recruited to date
Table 3 summarises key baseline attributes of the 1615 subjects recruited as at January 2021 according to current DMT class. The first patient was recruited to this study on 24 May 20. COVID-19-related delays substantially slowed rates of recruitment in 2020 and through the first half of 2021, from which time improvements have been seen. The low number of individuals with primary and secondary progressive MS is to be expected, given the inclusion criteria of the study and relatively recent approval of therapies for active progressive MS.
Ethics and dissemination
This study has had ethical review via London City and East REC (ref. 19/LO/0064). As this study uses routinely collected medical data as the primary data source, there are minimal burdens on participants.
As this is an observational study, the main risks are of data misuse and subject identification. This risk will be minimised by using double password protected computers, maintaining links between subject identifiers and an anonymised study code separate from the database in a locked compartment in a room with controlled access. Researchers with access to the data will agree not to attempt to identify individual subjects except in the context of direct responsibility for their clinical care. An oversight committee will be put in place to assess study conduct and data security on at least an annual basis, reporting to the study sponsor.
The OPTIMISE:MS study thus provides a unique opportunity for the UK to rise to the challenge of high-quality real world data studies in MS. It sets out to answer a vital research question, with a clearly defined core dataset. The use of an adaptable, disease-focussed platform for data entry enables the easy addition of add-on studies, such as those attempting to answer additional research questions, such as those around pregnancy, cognition or pharmacogenomics, subject to additional ethical approvals. While this study is a significant undertaking, it is one that is of clear benefit to people with MS—it is only by getting the systems and studies set up to the highest rigour that we will be able to provide clear answers regarding real life risks and benefits of treatment.
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @helenlouiseford
Contributors RD, MC and PMM conceived the study idea. RD drafted the initial study protocol with early and key input from MC and PMM. EW wrote the statistical analysis plan for the study protocol, and provided input into the remainder of the study design. AM, AC, SW, CH, JH, NE, NS, DR, RN, MM, CB, CAY and HLF reviewed, revised and contributed to the study protocol.
Funding Biogen IDEC Limited (grant ref P76049), Merck Serono UK Limited (grant ref WMCN_P74840) and Celgene Limited (Formally Bristol-Myers Squibb) (grant ref P76049) are providing the funding for this study.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.