Article Text

Abstract
Introduction Many preterm infants develop respiratory distress syndrome (RDS), a condition characterised by a relative lack of surfactant. Endotracheal surfactant therapy revolutionised the care of preterm infants in the 1990s. However, supporting newborns with RDS with continuous positive airway pressure (CPAP) and reserving endotracheal surfactant for those who develop respiratory failure despite CPAP yield better results than intubating all infants for surfactant. Half of preterm infants born before 29 weeks gestation initially managed with CPAP are intubated for surfactant. Intubation is difficult to learn and associated with adverse effects. Surfactant administration into the oropharynx has been reported in preterm animals and humans and may be effective. We wished to determine whether giving oropharyngeal surfactant at birth reduces the rate of endotracheal intubation for respiratory failure in preterm infants within 120 hours of birth.
Methods and analysis Prophylactic Oropharyngeal surfactant for Preterm infants: A Randomised Trial (POPART, Eudract No. 2016-004198-41) is an investigator-led, unblinded, multicentre, randomised, parallel group, controlled trial. Infants are eligible if born at a participating centre before 29 weeks gestational age (GA) and there is a plan to offer intensive care. Infants are excluded if they have major congenital anomalies. Infants are randomised at birth to treatment with oropharyngeal surfactant (120 mg vial <26 weeks GA stratum; 240 mg vial 26–28+6 weeks GA stratum) in addition to CPAP or CPAP alone. The primary outcome is intubation within 120 hours of birth, for bradycardia and/or apnoea despite respiratory support in the delivery room or respiratory failure in the intensive care unit. Secondary outcomes include incidence of mechanical ventilation, endotracheal surfactant use, chronic lung disease and death before hospital discharge.
Ethics and dissemination Approval for the study has been granted by the Research Ethics Committees at the National Maternity Hospital, Dublin, Ireland (EC31.2016) and at each participating site. The trial is being conducted at nine centres in six European countries. The study results will be submitted for publication in a peer-reviewed journal.
Trial registration number 2016-004198-41; Pre-results.
- neonatology
- paediatrics
- perinatology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first randomised study to specifically examine the efficacy of giving oropharyngeal surfactant at birth to preterm infants at high risk of developing respiratory distress syndrome (RDS).
We are enrolling infants <29 weeks gestation, including infants at 23 and 24 weeks gestation, who are most at risk of RDS.
We were unable to credibly mask the intervention.
To reduce the risk of bias, we used objective criteria for our primary outcome, that is, intubation within 120 hours of life.
The multicentre nature of this study will increase the generalisability of its findings.
Background
Respiratory distress syndrome (RDS) is a lung condition of the preterm infant. The risk of RDS is inversely related to gestational age (GA). Infants with RDS have structural and functional immaturity of their lungs. They also have a relative lack of surfactant,1 an endogenously produced substance that enables alveoli to expand more easily to recruit and maintain gas within the lung.
Exogenous surfactant2 3 is frequently used to treat newborns with RDS and has led to improved outcomes for infants worldwide. Randomised controlled trials (RCTs) in the 1980s–1990s4–7 demonstrated that surfactant, given by endotracheal tube (ETT), reduced mortality and air leak among premature infants who were intubated for respiratory failure due to RDS. This led to the widespread practice of intubating all extremely preterm infants for surfactant and ventilation (‘prophylactic surfactant’).5 Prior to the introduction of surfactant into clinical practice, concerns were raised that premature infants who were intubated for respiratory support had worse respiratory outcomes than infants who were managed with the non-invasive respiratory support, nasal continuous positive airway pressure (CPAP).8 These concerns persisted after the widespread introduction of surfactant.9 Multicentre RCTs found that starting infants on CPAP may be beneficial when compared with intubation and positive pressure ventilation (PPV); the studies reported decreased duration of mechanical ventilation with potential benefits of reduction of death and or bronchopulmonary dysplasia (BPD).10–12 Managing premature newborns initially with CPAP and reserving intubation, mechanical ventilation and surfactant for those infants with worsening respiratory failure despite CPAP yields better results than intubating all infants for surfactant administration.13 About half of the premature infants born before 29 weeks gestation initially managed with CPAP for RDS are ultimately intubated for surfactant and ventilation.14
Intubation is a procedure that is difficult to learn and is associated with adverse effects, both short15–19 and long term.20–22 This has led many clinicians to investigate alternative methods of surfactant delivery.23 Giving nebulised surfactant to spontaneously breathing infants has met with limited success.24–27 Progress has been slow due to the technical difficulties encountered in aerosolising such large molecules, the expense of the equipment needed to do so and the cost of the large amount of surfactant needed to form an aerosol. Interest has largely focused on less-invasive methods of surfactant administration.
Less-invasive surfactant administration (LISA) techniques involve introducing either a feeding tube or vascular catheter into the trachea of a spontaneously breathing infant at laryngoscopy.28–31 LISA is associated with lower rates of mechanical ventilation among preterm infants in randomised28 31 and observational studies.32 Two-year follow-up outcomes for infants enrolled in the randomised trial avoid mechanical ventilation,28 where infants were randomised to surfactant via LISA or to standard care with CPAP and ET instillation of surfactant if necessary, are similar between groups.33 The procedure is becoming more widely used, but rates vary between countries.34–37 Concerns regarding the validity and risk of bias within studies, a lack of familiarity with the technique, and patient discomfort have been reported as reasons for not using LISA.36 The use of sedation and analgesia prior to laryngoscopy is not standard for the LISA procedure,28 and the short-term adverse effects of laryngoscopy are not avoided. While meta-analyses report that the LISA technique is associated with less death or BPD,38–40 further RCTs are needed. The Optimist-A trial,41 evaluating minimally invasive surfactant therapy in preterm infants born between 25 and 28 weeks gestation, is ongoing.
The laryngeal mask airway, a supraglottic airway device, may be used as an interface to deliver surfactant.42–45 However, there is currently no device available for use in very low birth weight (BW) infants, who constitute the majority of infants diagnosed with RDS.
Direct administration of surfactant into the pharynx of human infants has been described in randomised7 and prospective cohort studies.46 47 It is apparently effective and is an easier technique to perform than endotracheal intubation or passing a feeding tube or vascular catheter into the trachea. Advantages of pharyngeal surfactant use are that it is an easy and cheaper method of administering surfactant and likely causes less discomfort to infants as it avoids the use of a laryngoscope. Giving surfactant early, prior to ventilation, delivers surfactant to a fluid-filled lung, which is spread via a fluid–air interface when the infant starts breathing. Animal studies report that surfactant is distributed more uniformly,48 and lung function and compliance is better49 if surfactant is delivered prior to ventilation. If shown to be effective, it may reduce the adverse effects, and additional associated costs, of ventilation.
A Cochrane review of pharyngeal surfactant50 did not identify any eligible trials to assess whether pharyngeal installation of surfactant before the first breath prevented morbidity and mortality in infants at risk of RDS. The Ten Centre Study randomised 328 infants born between 25 and 29 weeks gestation to artificial surfactant therapy or saline. For those randomised to surfactant therapy, the first dose was given via the oropharynx, with subsequent doses given via an ETT if the infant was intubated; however, the outcomes of infants who received pharyngeal surfactant alone were not reported. Large well-conducted RCTs are needed, due to the evidence from animal51 52 and observational human studies46 47 suggesting that pharyngeal surfactant administration is potentially safe, feasible and may be effective.
Objective
We are performing a study to establish whether giving preterm infants surfactant into their oropharynx at birth in addition to CPAP compared with CPAP alone reduces their need for subsequent intubation in the first 5 days of life.
Methods
Trial design
The Prophylactic Oropharyngeal surfactant for Preterm infants: A Randomised Trial (POPART) is an investigator-led, unblinded, multicentre, randomised parallel-group controlled trial. It aims to determine whether administering oropharyngeal surfactant to premature infants at birth in addition to CPAP compared with CPAP alone reduces the rate of intubation for respiratory failure in the first 5 days of life. The trial will recruit 250 infants born <29 weeks GA at participating centres. A schedule of events is seen in figure 1.
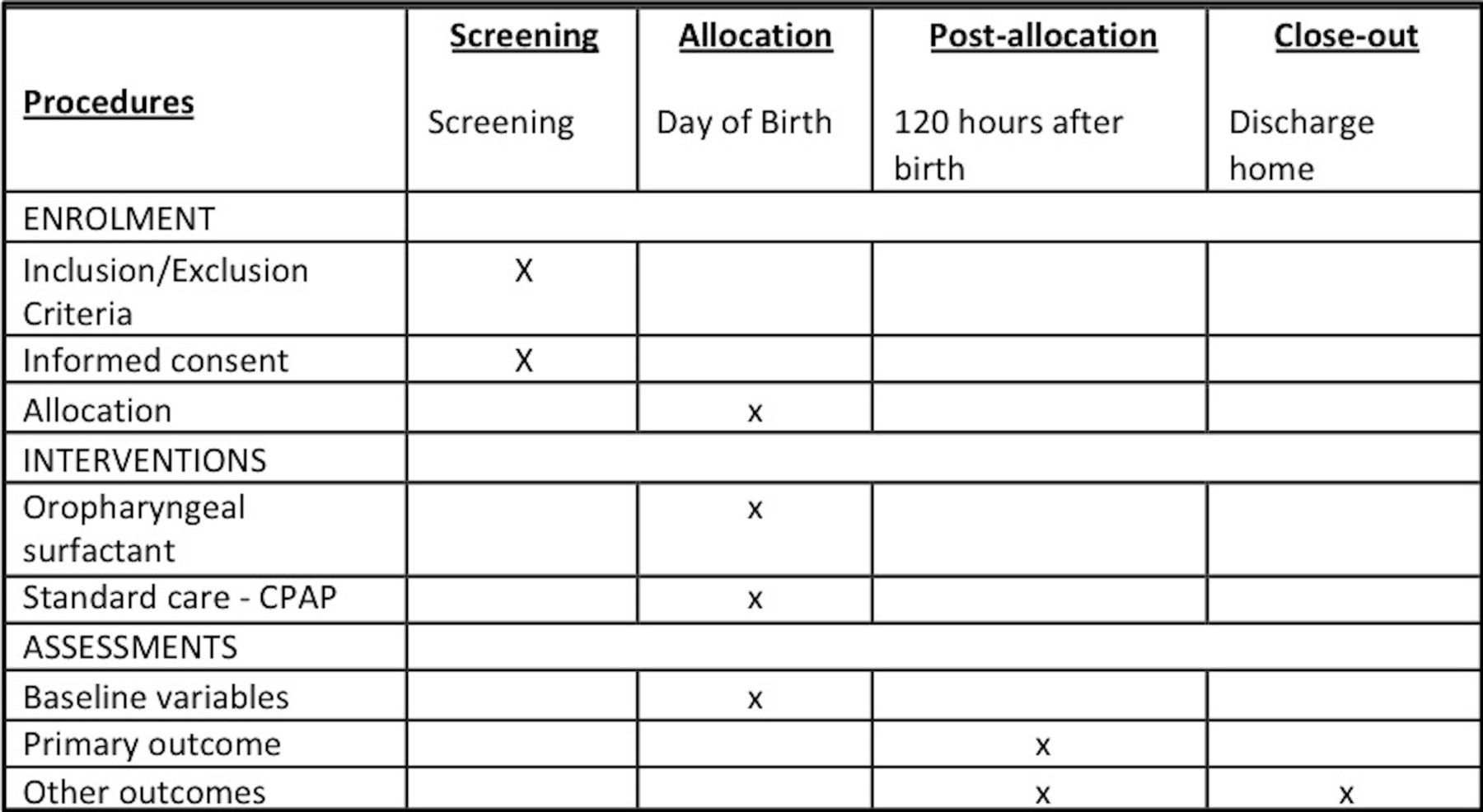
{kind=link}
Schedule of events. CPAP, continuous positive airway pressure.
Setting
The trial is being conducted at nine neonatal intensive care units (NICUs) in six European countries (Ireland (National Maternity Hospital (NMH), Dublin; Coombe Women and Infants University Hospital Dublin); Norway (University Northern Norway, Tromsø; Haukeland University Hospital, Bergen); Czech Republic (Charles University, Prague; University Hospital Brno, Brno); Belgium (Le Centre Hospitalier Universitaire, Liege); Sweden (Karolinska Institutet, Stockholm) and Portugal (Hospital de Braga, Braga)).
Participants
Inclusion and exclusion criteria
Trial subjects are premature infants at risk of RDS. Infants born <29 weeks GA are included if the treating physician plans to offer intensive care. Infants are excluded if they have major congenital anomalies (including neural tube defects, major structural cardiac anomalies (excluding patent ductus arteriosus (PDA), ventricular septal defect, atrioventricular septal defect), abdominal wall defects, congenital diaphragmatic hernia and major dysmorphic features with an abnormal karyotype) and if the treating physician does not plan to offer intensive care. If there is a known anomaly prenatally, families are not approached for consent. In the event of a postnatal diagnosis of the aforementioned conditions, these infants meet criteria for postrandomisation exclusion.
Written informed consent from parent/legal guardian(s) is obtained before delivery. Infants of multiple gestations and of either sex are eligible to be enrolled.
Screening and consent
Prior to the delivery a member of the research team or other senior doctor approaches parent(s)/guardian(s) of eligible infants to inform them about the study. The team member explains the purpose and nature of the study and provides written information for the parent(s)/guardian(s) to keep. If the local language is not their first language, they are offered the opportunity to have an interpreter present while the study is explained. Written consent for enrolment of the infant in the study is then sought. Parents are informed that they may withdraw their child from the study at any time should they so wish; and that a decision not to consent to their infants’ participation in the study or to withdraw their infant from the study once enrolled does not affect their infant’s access to the best available treatment and care.
Outcome measures
Primary outcome
The primary outcome is the incidence of endotracheal (ET) intubation for respiratory failure within 120 hours of birth. Enrolled infants are intubated for persistent apnoea and/or bradycardia (HR <100 bpm) in the delivery room (DR), or for respiratory failure in the NICU defined as ≥2 of:
Clinical signs—worsening tachypnoea; grunting; subcostal, intercostal and/or sternal recession.
Acidosis—pH <7.2 on two blood gases (arterial or capillary) ≥30 min apart.
Hypoxaemia—FiO2 >0.4 to keep oxygen saturation (SpO2) ≥90% for >30 min.
Hypercarbia—PCO2 >9.0 kPa on two blood gases (arterial or capillary) ≥30 min apart.
Apnoea—recurrent apnoea treated with mask ventilation.
The primary outcome is intubated within 120 hours of birth. For the purpose of the primary outcome, infants are recorded as ‘yes’ if they were intubated, briefly intubated for surfactant administration (for example, the "INtubtate-SURfactant-Extubate" or INSURE approach) and brief tracheal catheterisation for surfactant administration (for example, LISA technique).
We record the treatment plan at the time of intubation. We record whether there is (a) a plan for intubation with ETT, surfactant administration and continued ventilation; (b) a plan for ‘INSURE’—intubation with ETT, surfactant administration and immediate (<30 min) extubation; (c) a plan for surfactant administration using LISA technique—surfactant administration through a thin endotracheal catheter or (d) other.
We acknowledge that not all infants achieving ≥2 of the intubation indicators may be intubated.
Secondary outcomes
The secondary outcomes are as follows:
Intubation in the DR.
Number of attempts taken to successfully intubate in the DR.
Chest compressions in the DR.
Epinephrine administration in the DR.
Rectal temperature on admission to the NICU.
NICU intubation.
Surfactant use before death or hospital discharge.
Number of doses and total dose.
Intratracheal surfactant received post intervention.
Doses of postintervention surfactant.
RDS.
Clinical evidence and radiological evidence of respiratory distress at the time of first intubation.
Incidence of pneumothorax.
Incidence of pneumothorax on chest X-ray.
Pneumothorax treated with needle aspiration or chest drain insertion.
Incidence of pulmonary haemorrhage.
Clinical evidence of pulmonary haemorrhage.
Mechanical ventilation.
Days of mechanical ventilation.
Use of postnatal corticosteroids for ventilator dependence.
Days of duration of respiratory support (endotracheal ventilation, high-frequency oscillatory ventilation, CPAP, heated humidified high-flow nasal cannula O2 and low flow nasal cannula O2).
BPD—supplemental O2 at 28 days of life.
Chronic lung disease of prematurity (CLD)—O2 treatment at 36 weeks corrected GA; we are also recording physiological BPD as determined by physiological oxygen reduction test.
Medical treatment for a PDA.
Administration of ibuprofen or paracetamol for PDA.
Surgical treatment for a PDA.
Proven necrotising enterocolitis (≥Bell’s stage 2).
Incidence of intraventricular haemorrhage (IVH) (any and severe: IVH grade ≥3).
Evidence on surveillance cranial ultrasounds performed as standard of care.
Incidence of cystic periventricular leukomalacia.
Evidence on surveillance cranial ultrasounds performed as standard of care.
Retinopathy of prematurity treated with laser photocoagulation or intravitreal injections.
Evidence on surveillance ophthalmology review performed as standard of care.
Death before hospital discharge.
Survival without BPD at hospital discharge.
Survival without CLD at hospital discharge.
Duration of first hospitalisation.
Use of home oxygen therapy.
Discharged home on oxygen therapy.
Investigational medicinal product (IMP)
Poractant alfa (Curosurf, Chiesi Farmaceutici, Parma, Italy) is a natural surfactant prepared from porcine lungs. It is licensed for ET use and administration via thin catheter for the prevention and treatment of RDS in preterm infants. The dosing recommendations for treatment with Curosurf when given by ETT are 200 mg/kg for established RDS and 100–200 mg/kg for prophylaxis. Further doses of 100 mg/kg Curosurf may be given to infants who have persistent respiratory distress despite treatment with surfactant (maximum recommended dose 400 mg/kg). It is currently not licensed for oropharyngeal administration, and therefore this study is examining the off-label use of a licensed product. The timing or dosage of ET surfactant is not affected by oropharyngeal surfactant. If an infant is felt to need ET surfactant following initial oropharyngeal administration, then they receive the standard initial dose via ETT. Additional doses are given at the discretion of the attending physician.
Randomisation
Infants are randomised (1:1) to receive oropharyngeal surfactant in addition to CPAP or CPAP alone using variable block randomisation, with block sizes of 4, 6 and 8. Randomisation is stratified by participating centre and GA (<26 weeks and 26–28+6 weeks inclusive). Infants of multiple gestations are randomised as individuals.
A computer-generated randomisation schedule using sequential six-digit randomisation codes was prepared by an independent statistician who will not be involved with subsequent data analysis or interpretation and stored securely on a password-protected computer. Each participating centre is provided with separate boxes for the two GA strata with consecutively numbered, sealed opaque randomisation envelopes containing the assigned treatment allocation. The boxes containing the envelopes are stored securely in the NICU. An envelope from the appropriate box is opened immediately before birth.
Blinding
This is an open-label study. The study is not blinded to investigators, subjects, or medical or nursing staff. We are not using a placebo, and in the event of the infant being randomised to the ‘CONTROL’ group, then they will be commenced on CPAP immediately after birth. The trial statistician will be blinded for data analysis and will be kept unaware of treatment group assignments. We defined objective criteria for the primary outcome to minimise potential bias.
Intervention arm: oropharyngeal surfactant
Infants randomised to oropharyngeal surfactant receive a dose of poractant alfa (Curosurf, Chiesi Farmaceutici) immediately after birth, ideally before the cord is clamped (for example, at 60 s), and are then commenced on CPAP as per routine practice. If it is given after the cord is clamped, it is given once the infant is placed on the resuscitaire. It is given within 5 min of birth in all cases. We are recording the timing of cord clamping for all patients.
The surfactant is warmed prior to being drawn up in a sterile syringe as per the manufacturer’s recommendation. Surfactant is administered by opening the mouth gently and giving the surfactant as a single bolus into the oropharynx using a thin flexible catheter attached to the syringe.
Infants are not weighed prior to enrolment. The 50th centile for BW for boys and girls according to GA is shown in table 1. In our study, infants <26 weeks receive a full 120 mg vial of Curosurf. We estimate that this provides dosing in the range as indicated in table 2. In our study, infants 26–28 weeks receive a full 240 mg vial of Curosurf, and we estimate that this provides dosing in the range as indicated in table 3.
50th centile for birth weight (BW) for boys and girls according to gestational age (GA)
Infants <26 weeks estimated dosing range, following 120 mg vial of Curosurf
Infants 26–28+6 weeks estimating dosing range, following 240 mg vial Curosurf
Control group: CPAP
Infants randomised to the control group do not have anything injected into their oropharynx and are stabilised on CPAP in the DR as per routine practice.
Clinical management
After the initial intervention, infants then receive standard care with CPAP, regardless of their group assignment. DR care is carried out by the neonatal team who are trained in neonatal resuscitation as per the recommendations of the International Liaison Committee on Resuscitation (ILCOR). Infants in both groups are intubated in the DR for persistent apnoea and/or bradycardia despite PPV by mask as per the ILCOR recommendations. Infants are not intubated in the DR solely for surfactant administration. All other aspects of neonatal intensive care are at the discretion of the treating physicians. Infants in both groups are treated equally. The frequency of blood gas monitoring is based on the decision of the treating physician. Enrolled infants are intubated if they reach the predetermined criteria for respiratory failure. After giving endotracheal surfactant for the treatment of RDS, attending clinicians may attempt to extubate the babies immediately or they may elect to ventilate the babies for a longer period at their discretion.
Data management
Data are collected by the on-site investigators from the patient’s clinical notes. This is recorded on a data worksheet and transferred to an electronic case report form (CRF) to be stored in a secure, dedicated, password-protected electronic database. The clinical study monitor and representative of the regulatory authority can directly access source documents for comparison of such data with the data in the electronic CRFs and can verify that the study is carried out in compliance with the protocol and local regulatory requirements.
The investigators adheres to national and hospital protocols on data use and storage. Data are coded. These data are stored in a locked filing cabinet then uploaded onto a password-protected computer in a locked office. Documents are stored safely in confidential conditions. On all study-specific documents other than the signed consent, the subject is referred to by the study subject identification code.
Description of statistical methods
Trial results will be reported according to the Consolidated Standards of Reporting Trials (CONSORT). The flow of patients through the trial will be represented on a CONSORT flow diagram, and the number included in the primary and secondary analyses as well as all reasons for exclusions will be reported per trial arm. Analysis of efficacy endpoints will be carried-out following the intention-to-treat principle. A per-protocol analysis will also be carried out on the primary endpoint, excluding infants with incomplete data on the primary outcome and infants with any major protocol deviations.
Demographic and baseline data will be summarised by treatment group to evaluate comparability.
Primary outcome analysis
The primary outcome will be summarised per group. The ratios of relative risk will be reported with 95% CIs. A two-sided, two-proportion Z test will be carried out to investigate whether the rate of endotracheal intubation differs between intervention and standard of care. This analysis will be carried out both on the intention-to-treat set and on the per-protocol set.
A completing risks model will be fitted to investigate the effect of the intervention on the primary endpoint, adjusting for competing outcomes (eg, mortality) that may impact on observation of the primary endpoint.
The sensitivity of the estimated intervention effect to measured covariates of interest, including centre, GA, BW, gender, mode of delivery and antenatal corticosteroid treatment, will be evaluated with regression analysis.
Secondary outcome analysis
Categorical outcomes will be summarised per treatment group, with between-group differences expressed as a relative risk with 95% CIs. A two-sided, two-proportion Z test will be carried out for each categorical outcome to investigate whether the proportion differs between intervention and standard of care. For the important secondary endpoint of death before hospital discharge, regression analysis will be employed to determine sensitivity of the estimated intervention effect to potentially relevant covariates (as specified above for the primary outcome).
Numeric secondary outcomes will be summarised by treatment group and between-group differences will be presented with a 95% CI. A superiority hypothesis test will be carried out to test for a difference in the outcome between control and intervention, using a t-test or a Mann-Whitney U test where relevant.
Subgroup analyses
Subgroups of interest include infants of different GA strata (eg, <26 weeks, and 26–28 weeks gestation at birth), and infants from different participating centres. Subgroup analysis of the primary outcome and the important secondary outcome of death before hospital discharge will be carried out by regression modelling to determine differences in the intervention effect for infants of different GA strata, and infants from different participating centres.
Missing data
Any missing data or data anomalies will be communicated to the study site(s) for prompt clarification and resolution. For outcomes missing more than 5% of data in either treatment group, missing data methods will be employed in analysis. For categorical outcomes with censored data, Kaplan-Meier analyses will be used to estimate treatment effect. For other missing data, a suitable imputation method will be selected during blind review of the data.
Sample size and power
The sample size calculation assumed a rate of endotracheal intubation of 46% for infants treated with CPAP alone, and a rate of 28% for infants receiving oropharyngeal surfactant and CPAP. The former was informed by published RCTs showing a rate of mechanical ventilation in the days after birth among preterm infants treated with CPAP alone from 40% to 60%10–12 and rates of CPAP failure of 43% reported in a cohort of preterm infants 25–28 weeks gestation initially commenced on CPAP.14 The latter was informed by a cohort of infants born 26–28 weeks gestation reporting that minimally invasive surfactant techniques reduced the rate of mechanical ventilation from 46% to 28%.28 Sample size was calculated in G*power based on a two-sided, two-proportion Z test. A sample size of 125 infants per arm will be required to give a statistical power of 80% at a significance level of 5%, adjusted for an anticipated death rate of 10% (estimated from local data (NMH, Neonatal Clinical Report, 2015)).
Safety analyses
Adverse events following administration of oropharyngeal surfactant will be documented. Safety analyses will be carried out on the Safety Set, defined as patients in the intervention arm who received oropharyngeal surfactant and patients who received CPAP only. The frequency of adverse events and the number and percentage of infants reported as having at least one emergent adverse event, will be reported by system organ class and preferred term, by treatment received. The same description will be performed for serious adverse events (SAE), severe AE, AE treatment related and AE leading to IMP withdrawal. Defined SAEs for the study are important medical events and death before hospital discharge.
Safety monitoring and interim analysis
A data safety monitoring board (DSMB) will be established to perform ongoing safety surveillance and to perform interim analyses on the study data. The DSMB will be an independent committee, composed of a minimum of three members; at least two will be clinicians with expertise in clinical trials; at least one member will be a clinician with expertise in neonatology. They will not be blinded to the intervention groups.
The DSMB will meet on a 6 monthly basis after start of the trial and will review the frequency and severity of AEs in both treatment groups. If they observe any significant excess of SAEs in the intervention group associated with the intervention, they may recommend premature termination of the trial on the basis of safety concerns.
The DSMB will conduct interim analysis to determine whether the data provide overwhelming evidence of efficacy or futility, defined as a highly statistically significant difference in the primary outcome or a highly statistically significant difference in the important secondary outcome of death before hospital discharge. The type I error rate for interim analysis will be set to 0.001 in accordance with the Haybittle-Peto stopping boundary. For final analysis, the type I error rate will remain at 0.05. Interim analysis will be carried out after approximately 50% of participants (n=126) have completed the study. The DSMB may recommend early termination of the trial due to efficacy or futility; or for unanticipated concerns for the safety of enrolled infants. Standard procedures for reporting AEs will be used in accordance with Good Clinical Practice guidelines.
Ethics and dissemination
The study was initially approved by the Research Ethics Committee at NMH, Dublin, and the Health Products Regulatory Authority of Ireland. Approval was also obtained at the research ethics committees at each participating site and at the relevant competent authority for each participating country. All bodies are informed in writing of any substantial changes to the protocol, prior to any such changes being implemented. University College Dublin, Ireland, is the sponsor for this study. The study protocol was registered at the EU Clinical Trials Register before the first patient was enrolled (Eudract No. 2016-004198-41)
Patient and public involvement
We liaised with the Irish Neonatal Health Alliance for assistance when designing the parent information leaflet and consent form. Parent focus groups were held via Pediatric Clinical Research Infrastructure Network prior to expansion of the study to European sites.
Recruitment
The NMH is a stand-alone university maternity hospital with a tertiary NICU to which >150 infants <1500 g are admitted annually. Approximately 60 babies <29 weeks gestation are admitted annually. Though the enrolment rates to our studies among eligible infants are consistently excellent (>80%), we believe it is necessary to enrol infants at multiple sites in order to enrol our planned target sample of 250 infants in a timely fashion. We have a track record enlisting the help of collaborators nationally53 and internationally54 55 to perform our studies. We believe that with their help, we can enrol these infants in 3 years.
Current status
The trial began recruitment in December 2017, with additional sites joining subsequently. It is currently recruiting in nine centres in six European countries. It is expected that recruitment for the study will be completed by December 2020.
Publication of results
The authors intend to publish the results of this trial in a high-quality, peer-reviewed journal on completion of data collection and analysis.
Discussion
Oropharyngeal surfactant given immediately after birth to preterm infants at risk of RDS has the potential to reduce the risk of intubation and ventilation. Endotracheal intubation is invasive and unpleasant for newborns that is associated with adverse short-term and long-term effects. It is also a skill that is difficult for clinicians to learn and maintain. In contrast, giving surfactant into the oropharynx is easy and avoids the adverse effects associated with intubation. There is evidence from animal studies and from case series in humans that it may be effective. This is an attractive proposition, because it could avoid harms associated with intubation for babies and raises the possibility of giving surfactant in contexts where it is not currently feasible (eg, non-tertiary settings, developing countries). We were unable to credibly mask the intervention and acknowledge this lack of blinding as a limitation of the study. We tried to minimise potential bias by setting predefined objective treatment failure criteria, which were agreed on by all participating sites.
Acknowledgments
We would like to acknowledge the administrative staff at UCD for their advice, and support with the trial thus far. We wish to thank the members of Pediatric Clinical Research Infrastructure Network and the principal investigators and members of the Clinical Trials Units at all our European sites for facilitating extension of our trial to Europe. Prophylactic Oropharyngeal surfactant for Preterm infants: A Randomised Trial Investigators: Jan Miletin, Coombe Women and Infants University Hospital, Dublin, Ireland; Claus Klingenberg, University Northern Norway, Tromsø, Norway; Hans Jørgen Guthe, Haukeland University Hospital, Bergen, Norway; Richard Plavka, Charles University, Prague, Czech Republic; Tomas Juren, University Hospital Brno, Brno, Czech Republic; Vincent Rigo, Le Centre Hospitalier Universitaire, Liege, Belgium; Kajsa Bohlin, Karolinska Institutet, Stockholm, Sweden; and Almerinda Pereira, Hospital de Braga, Braga, Portugal.
References
Footnotes
Contributors CO conceived and designed the trial protocol, co-wrote the first draft and revised the manuscript for intellectual content. MCM helped design the trial protocol, co-wrote the first draft and revised the manuscript for intellectual content. MG, BM, RH and PD helped design the trial protocol and revised the manuscript for intellectual content. MG designed the statistical analysis plan. All authors approved the final manuscript prior to submission.
Funding Chiesi Farmaceutici, manufacturers of poractant alfa (Curosurf), supply the study drug free of charge; they had no role in study design; have no role in study conduct; and will have no role in data collection, analysis or interpretation. Further, they will have no role in the decision to present, publish or otherwise report results.Madeleine Murphy is the recipient of a Clinical Research Fellowship by the National Children’s Research Centre, Dublin (R17637) Pediatric Clinical Research Infrastructure Network (PedCRIN), a Project of the European Union Horizon 2020 programme designed to support the extension of clinical drug trials in children throughout Europe, also support the study.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.