Article Text

Abstract
Introduction Acute kidney injury (AKI) is a frequent complication after cardiac surgery with adverse short-term and long-term outcomes. Although prevention of AKI (PrevAKI) is strongly recommended, the optimal strategy is uncertain. The Kidney Disease: Improving Global Outcomes (KDIGO) guideline recommended a bundle of supportive measures in high-risk patients. In a single-centre trial, we recently demonstrated that the strict implementation of the KDIGO bundle significantly reduced the occurrence of AKI after cardiac surgery. In this feasibility study, we aim to evaluate whether the study protocol can be implemented in a multicentre setting in preparation for a large multicentre trial.
Methods and analysis We plan to conduct a prospective, observational survey followed by a randomised controlled, multicentre, multinational clinical trial including 280 patients undergoing cardiac surgery with cardiopulmonary bypass. The purpose of the observational survey is to explore the adherence to the KDIGO recommendations in routine clinical practice. The second phase is a randomised controlled trial. The objective is to investigate whether the trial protocol is implementable in a large multicentre, multinational setting. The primary endpoint of the interventional part is the compliance rate with the protocol. Secondary endpoints include the occurrence of any AKI and moderate/severe AKI as defined by the KDIGO criteria within 72 hours after surgery, renal recovery at day 90, use of renal replacement therapy (RRT) and mortality at days 30, 60 and 90, the combined endpoint major adverse kidney events consisting of persistent renal dysfunction, RRT and mortality at day 90 and safety outcomes.
Ethics and dissemination The PrevAKI multicentre study has been approved by the leading Research Ethics Committee of the University of Münster and the respective Research Ethics Committee at each participating site. The results will be used to design a large, definitive trial.
Trial registration number NCT03244514.
- acute renal failure
- cardiac surgery
- adult intensive & critical care
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This will be the first multinational trial using a biomarker-guided approach to detect high-risk patients for acute kidney injury (AKI).
The strength of the prevention of AKI (PrevAKI) multicentre project is the combination of a survey with a multicentre-randomised controlled trial to explore routine clinical practice and to investigate the feasibility of the study protocol in multiple centres.
The PrevAKI multicentre trial is not powered to evaluate the preventive effect of the Kidney Disease: Improving Global Outcomes bundles on the occurrence of AKI and therefore a definitive future trial will be needed.
Introduction
Acute kidney injury (AKI) is a well-recognised complication after cardiac surgery.1 Depending on the definition used, AKI occurs in up to 45% of cardiac surgery patients, and approximately 1%–2% of patients who require renal replacement therapy (RRT).2–4 The underlying mechanisms of cardiac surgery-associated AKI are not fully understood, but ischaemia-reperfusion injury, inflammation and tubular epithelial cell dysfunction often contribute.5
Independent of the underlying aetiology, AKI is associated with increased morbidity and mortality, especially in patients undergoing cardiac surgery with cardiopulmonary bypass (CPB).6 7 Although most patients develop mild AKI, mortality rates in these patients are five times higher compared with patients without AKI.8 Moreover, patients who survive an episode of severe AKI are at high risk of developing chronic kidney disease (CKD), which is associated with a worse long-term outcome and a tremendous economic burden for the healthcare system.9 Therefore, prevention of AKI has a high priority.10 Despite numerous clinical trials using different pharmacological treatments, the optimal strategy to prevent AKI is unknown.
The Kidney Disease: Improving Global Outcomes (KDIGO) guideline from 2012 includes various recommendations to prevent AKI in high-risk patients, including the discontinuation of all nephrotoxic agents when possible, optimisation of volume status and haemodynamics, consideration of a functional haemodynamic monitoring, close monitoring of serum creatinine and urine output, avoidance of hyperglycaemia and consideration of alternatives to radiocontrast agents.11
Investigations have revealed that adherence with treatment bundles is often low in routine clinical practice.12 In addition, treatment bundles need to be applied before the condition of interest actually develops. For AKI, this means that the KDIGO recommendations should be implemented in high-risk patients before the onset of AKI. Novel biomarkers have been shown to identify patients at high risk for AKI. Although a variety of biomarkers can predict AKI after cardiac surgery,13 point-of-care devices to measure biomarkers at the bedside are only for neutrophil gelatinase-associated lipocalin (NGAL), and tissue inhibitor of metalloproteinases-2 and insulin-like growth factor-binding protein7 ([TIMP-2]*[IGFBP7]). NGAL is influenced by different comorbidities whereas [TIMP-2]*[IGFBP7] has a very high specificity and very good predictive value. Therefore, we decided to use [TIMP-2]*[IGFBP7] to identify patients at increased risk for AKI.14–16
In a single-centre trial, we previously demonstrated that biomarker-guided implementation of the cardiovascular (CV) surgery AKI bundle (derived from the KDIGO bundle) significantly decreased the occurrence of AKI in high-risk patients undergoing cardiac surgery (55.1% intervention vs 71.7% control group; p=0.004).17 In preparation for a definitive adequately powered future randomised controlled trial (RCT), we designed the prevention of acute kidney injury (PrevAKI) multicentre feasibility study to test the protocol in a multicentre setting.
Objectives and aims
Aim 1 (observational study)
To evaluate adherence with the KDIGO guidelines in routine clinical practice, we will test the following hypothesis:
Hypothesis I: the CV surgery AKI bundle is not applied to all high-risk patients undergoing cardiac surgery.
Aim 2 (interventional trial)
To demonstrate that the implementation of the CV surgery AKI bundle in cardiac surgery patients at high risk for AKI is feasible in a multicentre trial, we will test the following hypothesis:
Hypothesis II: it is possible to implement the CV surgery AKI bundle in high-risk patients undergoing cardiac surgery in the context of a multicentre study.
Methods and analysis
Design and setting
This study is a prospective, observational survey followed by a randomised-controlled, parallel group multicentre trial conducted in 12 centres across Europe including the UK (online supplementary eTable S1). The concept of the study was approved by all members of the steering committee (online supplementary eTable S2). The protocol follows Consolidated Standards of Reporting Trials and the conduct of the study is in accordance with the Declaration of Helsinki (version Fortaleza, 2013). The flow chart is summarised in figure 1.
Supplemental material

Trial workflow. Patients will be screened in all participating sites for eligibility on a daily basis. Prior to enrolment, it is assured that none of the exclusion criteria are present and informed consent process is performed. Four hours after cardiopulmonary bypass (CPB), urine samples for the measurement of tissue inhibitor of metalloproteinases-2 and insulin-like growth factor-binding protein 7 [TIMP-2]*[IGFBP7] will be collected. In case [TIMP-2]*[IGFBP7] is ≥0.3, patients are at high risk and randomisation can be performed. Patients in the intervention group will receive a functional haemodynamic monitoring (according to the routine of each centre) immediately after randomisation and haemodynamic situation will be optimised according to a haemodynamic algorithm (figure 2). Laboratory test will be analysed and variables relevant for the assessment of renal and haemodynamic situation and safety will be recorded. Follow-up will be performed after 30, 60 and 90 days. ACEi, ACE inhibitors; AKI, acute kidney injury; ARBs, angiotensin receptor blockers; eGFR, estimated glomerular filtration rate; HES, hydroxyethyl starch; KDIGO, Kidney Disease: Improving Global Outcomes; RRT, renal replacement therapy; SCr, serum creatinine.
Patient and public involvement
In Germany, Italy, Spain and Belgium, patients and public were not involved in the research design of the study. In the UK, members of the Kidney Patient Association reviewed the protocol and were involved in designing the patient information sheets. Study results will be published open access. At request, patients or their representatives will be informed about the results of the trial.
Participants
In the survey, we will include all adult patients undergoing cardiac surgery with CPB on two consecutive days in all participating centres.
In the interventional trial, patients need to meet all eligibility criteria to participate in the trial. All four inclusion criteria need to be fulfilled prior to randomisation: (1) patients undergoing cardiac surgery with CPB, (2) urinary [TIMP-2]*[IGFBP7]≥0.3 at 4 hours after CPB, (3) age between 18 and 90 years and (4) written informed consent. Patients are excluded if any of the following criteria is present: pre-existing AKI (≥stage 1), need for cardiac assist devices (extracorporeal membrane oxygenation, left ventricular assist device, right ventricular assist device, intra-aortic balloon pump), pregnancy or breast feeding, known glomerulonephritis/interstitial nephritis/vasculitis, CKD with an estimated glomerular filtration rate <20 mL/min/1.73 m2, chronic dialysis dependency, prior kidney transplant within the last 12 months and participation in another interventional trial within the last 3 months. In addition, patients with any kind of relationship with the investigator or employed by the sponsor/investigator and patients held in an institution by legal or official order will not be included in the trial.
Consent process
For the survey, written informed consent will be obtained according to the country-specific requirement of the research ethics committee.
For the interventional trial, a member of the research team will inform the patient about the nature of the trial, its aims, potential benefits as well as possible risks (online supplementary material 1). Participants will be asked to give written consent to participate in the study prior to surgery. The consent form will be countersigned by a member of the research team. The original document will be stored in a secure place and the patient will receive a copy. If the patient is unable to provide written consent, a legally authorised representative will be consulted. If an authorised representative is not immediately available, the research team will follow the agreed recommendations of their local research ethics committee or country-specific institutional review boards.
Supplemental material
All data will be kept confidential and will be stored in pseudonymised form. The patient has the right to withdraw from the study at any time without providing a reason and without their medical treatment being affected.
Observation
In the observational part, all adult patients undergoing cardiac surgery in the participating centres will be included. Data regarding routine management will be collected, especially the adherence with the different elements of the KDIGO bundle (online supplementary material 2). This includes (1) discontinuation of all nephrotoxic agents, (2) optimisation of volume status and haemodynamics (target mean arterial pressure (MAP) ≥65 mm Hg (not fulfilled if MAP is <65 mm Hg and no treatment is initiated to achieve this goal), (3) functional haemodynamic monitoring (including echocardiography, pulse contour cardiac output or pulmonary artery catheter), (4) close monitoring of serum creatinine every 12 hours and urine output hourly, (5) avoidance of hyperglycaemia (blood glucose levels ≤150 mg/dL or ≤8.3 mmol/L) and (6) avoidance of radiocontrast agents.
Supplemental material
Randomisation and blinding
In the interventional trial, randomisation will be performed centrally using a web-based randomisation system provided by the European Society of Intensive Care Medicine. Randomisation lists will be stratified by centre, and generated using block randomisation with randomly varying block sizes. Randomisation assignment (in a 1:1 ratio to the two treatment arms) will be given only to those patients who fulfil the inclusion criteria and have none of the exclusion criteria, and where informed consent has been obtained. The treatment protocol will be applied immediately after randomisation.
Blinding of the intervention is not possible. Study intervention will be performed by clinical staff involved in anaesthesia and perioperative care. Endpoint assessment will be performed by a blinded researcher not involved in providing anaesthesia and perioperative care. Patients will remain blinded to allocation.
Treatment
In the survey, there will be no study-related treatment procedures. Every patient will be treated according to the standards at the individual centre.
In the interventional trial, 4 hours after CPB, urinary [TIMP-2]*[IGFBP7] will be measured to identify patients at high risk for AKI. Patients with a value ≥0.3 (ng/mL)2/1000 will be randomised to one of the two treatment arms.
Control group: patients will receive standard of care as per centre.
Intervention group: the CV surgery AKI bundle will be strictly implemented.
At different time points, blood and urine samples from recruited patients will be collected to analyse different biomarkers. All other therapies will be directed by the patient’s clinical team. All medications will be dose adjusted according to renal function in accordance with standard dosing guidelines.
CV surgery AKI bundle
The CV surgery AKI bundle includes the following measures:
Discontinuation of all nephrotoxic agents if possible.
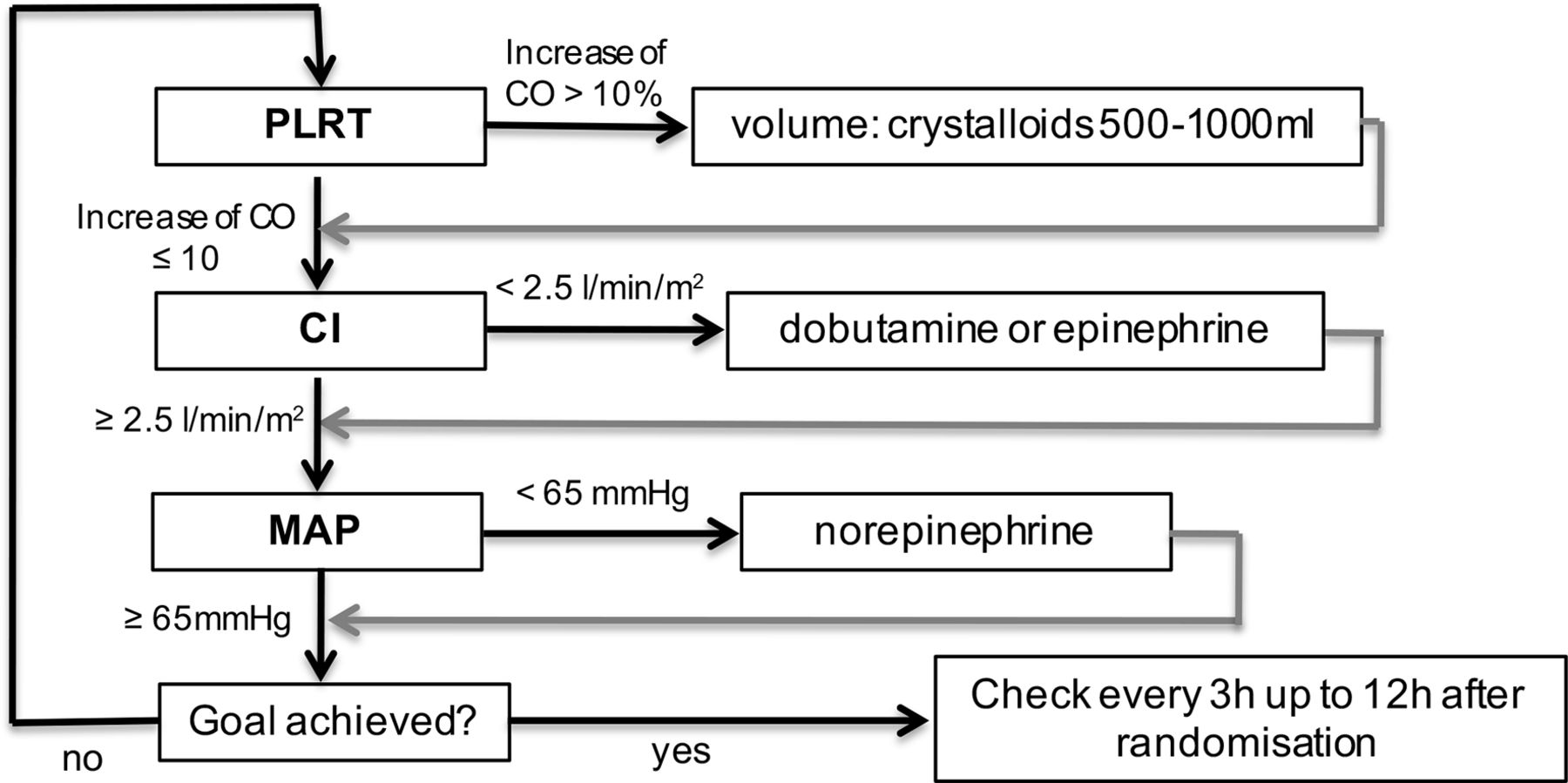
Haemodynamic monitoring and optimisation according to a predefined haemodynamic algorithm (figure 2).
Close monitoring of serum creatinine (twice a day), fluid balance (twice a day) and urine output (every hour as long as the patient has a Foley catheter; if the patient does not have a Foley catheter, the urine is collected for a certain time period and divided by the number of hours to calculate hourly urine volume).
Avoidance of hyperglycaemia (defined as >150 mg/dL for more than 3 hours) by close monitoring for the first 72 hours after surgery.
Consideration of alternatives to radio contrast agents.
Discontinuation of ACE inhibitors and angiotensin II receptor blockers for the first 48 hours after surgery.
Avoidance of hydroxyethyl starch, gelatin and chloride-rich solutions.
{kind=link}
{kind=link}
Haemodynamic algorithm of the intervention group. Patients of the intervention group will receive a functional haemodynamic monitoring and an optimisation according to a haemodynamic algorithm consisting of three steps which need to be checked every 3 hours: step 1—performance of the passive leg raising test. If the cardiac output (CO) increases by >10%, then volume has to be supplemented (crystalloids 500–1000 mL according to the treating intensivist). If CO is ≤10%, step 2—measurement of the cardiac index (CI). If the CI is <2.5 L/min/m2, then dobutamine or epinephrine (according to the treating intensivist) needs to be applied. If CI is >2.5 L/min/m2, then step 3—measurement of mean arterial pressure (MAP). If the MAP is <65 mm Hg, then norepinephrine needs to be adjusted. If MAP is >65 mm Hg, then the goal is achieved. This is checked every 3 hours up to 12 hours after randomisation. PLRT, passive leg raising test.
Outcomes
In the survey, we will explore whether the different elements of the KDIGO bundle are already incorporated into routine clinical practice in the different centres. The primary endpoint of this observation is the binary indicator whether the CV surgery AKI bundle is completely fulfilled at all times or not.
Secondary outcomes will include
adherence to the different components of the KDIGO bundle.
The primary outcome of the interventional trial is the compliance rate defined as proportion of patients who are treated according to the trial protocol (CV surgery AKI bundle fulfilled at all time). Adherence to the trial protocol is achieved if all elements of the bundle are followed. In addition, we will calculate the adherence with the different individual components of the bundle.
Secondary outcomes include:
Occurrence of AKI within the first 72 hours after cardiac surgery (AKI as defined by the KDIGO criteria).
Moderate and severe AKI within 72 hours (according to the KDIGO criteria).
Free days through day 28 of vasoactive medications and mechanical ventilation.
Renal recovery (defined as serum creatinine <0.5 mg/dL higher than baseline) at days 30, 60 and 90.
All-cause mortality at days 30, 60 and 90.
Length of stay in intensive care unit and hospital.
Use of RRT at days 30, 60 and 90.
Major adverse kidney events (MAKE90 consisting of mortality, dialysis dependency persistent renal dysfunction at day 90).
Determination of different biomarkers (eg, TIMP2, IGFBP7, NGAL, kidney injury molecule-1, chitinase, hyaluronic acid).
Complications related to the implementation of the KDIGO guidelines documented (eg, arrhythmias).
Sample size
For the survey, no power analysis is required. The number of patients is based on surgical volumes in the two consecutive days prior to the interventional trial and is expected to be about n=100.
In the interventional trial, 1000 patients will be assessed for eligibility, and 280 patients will be enrolled into the trial and randomised (based on our previous findings). This sample size is determined as follows:
Patients will be randomised in a 1:1 ratio to one of the two treatment arms (intervention vs control). Randomisation serves to estimate the incidence of AKI as a secondary outcome to generate preinformation in preparation for an upcoming confirmatory trial.
The primary endpoint of the interventional trial is the compliance rate, that is, the proportion of patients treated according to the trial protocol (intervention group: CV surgery AKI bundle fulfilled at all time; control group: documented standard of care according to current clinical practice). In order to estimate the compliance rate, the two treatment groups will be pooled together. In the primary analysis, the compliance rate across all study patients will be estimated along with a two-sided 95% CI.
As the compliance rate cannot be quantified in advance, we pursue a worst-case approach and assume a compliance rate of 50%. In this case, the CI has maximal width. Based on this assumption, with a sample size of n=280 patients, the calculated 95% CI according to Clopper-Pearson (Fleiss 2003) ranges from 44% to 56%. Therefore, in case of any observed compliance rate apart from the worst-case scenario, the 95% CI is never wider than 56%−44%=12%. This corresponds to an estimation of the compliance rate with a precision of ±6%.
Statistical analysis
The study is designed to examine the feasibility of the trial protocol in a multicentre setting (as previously described).
In the observational study, we will evaluate whether the different elements of the KDIGO guidelines are already implemented in routine clinical practice. The primary endpoint of the observational study is the binary indicator whether the CV surgery AKI bundle is completely fulfilled at all times or not. Descriptive data analyses will include the calculation of means, SD and absolute and relative frequencies. No formal statistical hypothesis will be tested.
In the interventional trial, beyond descriptive analyses, inferential statistical analyses will be performed. In the primary statistical analysis, the two treatment groups will be pooled together and the compliance rate across all recruited study patients will be estimated along with a two-sided 95% CI according to Clopper-Pearson (Fleiss 2003).
Randomisation serves to estimate the incidence of AKI separately in two treatment groups as secondary outcome, in order to generate preinformation in preparation for an upcoming confirmatory trial. Randomisation will be checked by suitable two-sided statistical tests (χ2, or Fisher’s exact test for categorical data, Students’ t-test or Mann-Whitney U tests for continuous data). If normality of the data is not given, non-parametric methods will be used. Secondary endpoints will be analysed in the intention to treat collective using Fisher’s exact tests or χ2 tests for categorical data, and Student’s t-tests or Mann-Whitney U tests for continuous data. In a secondary analysis, we will investigate whether there is a difference in the incidence of AKI as defined by serum creatinine, urine output and the composite of the two between the treatment groups. Prognostic risk factors for AKI will be identified using a multivariable logistic regression analysis, accounting for confounding factors. In particular, based on a full model with potentially relevant covariates (eg, sex, age, previous heart surgery, preoperative creatinine), the final model will be established applying a stepwise variable selection procedure.
Ethics and dissemination
The PrevAKI-multicentre study has been approved by the leading Research Ethics Committee of the University of Münster (2017-291 f-S) and the corresponding Research Ethics Committee at each participating site (online supplementary eTable S3). Trial oversight will be performed by an independent Data Safety and Monitoring Board. The task is to oversee the safety of the trial subjects and to monitor integrity and validity of the conduct of the clinical trial.
The results will be presented at national as well as international conferences. The final manuscript will be published in a peer-reviewed journal and results will be used to design a large multicentre trial with the primary objective to investigate whether the implementation of the CV surgery AKI bundle reduces the occurrence of AKI in high-risk patients undergoing cardiac surgery.
The Standard Protocol Items: Recommendations for Interventional Trials checklist is included in the online supplementary material.
Supplemental material
Discussion
The PrevAKI-multicentre study is a prospective, observational survey followed by randomised controlled, multicentre and multinational, clinical trial with the primary goal to investigate the feasibility of the study protocol in a multicentre and multinational setting. In the absence of any preventive pharmacological options, the KDIGO guidelines recommend a bundle of supportive measures in patients at high risk for AKI.11 Whether these recommendations truly reduce the occurrence of AKI remains to be confirmed. In the first single-centre randomised controlled PrevAKI trial, we could demonstrate that the new renal biomarkers [TIMP-2]*[IGFBP7] identified patients at high risk for the development of AKI and that the CV surgery AKI bundle significantly reduced the occurrence of AKI by 15%.17 A further single-centre trial demonstrated similar results in patients undergoing major abdominal surgeries receiving a bundled intervention.18 However, these were single-centre trials and single-centre trials often show positive results. A large RCT is needed to confirm these findings. In preparation, it is necessary to explore first how many patients are already treated according to the KDIGO recommendations and second whether the implementation of the CV surgery AKI bundle is feasible in a multicentre, multinational setting. The results will inform the researchers whether changes to the protocol are required before proceeding with a large trial to reduce the occurrence of AKI after cardiac surgery.
Trial status
Recruitment started in November 2017. We estimate to complete the study in January 2020.
Acknowledgments
The authors would like to thank all the participating centres and corresponding staff member involved in this trial.
References
Footnotes
MK and CM are joint first authors.
MK and CM contributed equally.
MM and AZ contributed equally.
Contributors MK, CM, RW, SC, NGN, GT, MH, CA, HW, MI, FM, ADP, MGA, SI, VCS, GK, SN, CL, WV, PMH, PG, RW performed the study and drafted the manuscript. MO and EAJH helped designing the trial, performed the study and drafted the manuscript. JK, LF and JG helped designing the trial and drafted the manuscript. CW helped designing the trial, performed study coordination and drafted the manuscript. MM and AZ conceived the study, designed the trial and drafted the manuscript. All authors read and approved the final manuscript. MM and AZ share last authorship.
Funding The trial is supported by the European Society of Intensive Care Medicine.
Disclaimer The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the funding agency.
Competing interests MM, JK and AZ have received lecture fees from Astute Medical, Fresenius and Baxter, unrelated to the current study. JK and AZ have received grant support from Astute Medical, unrelated to the current study. MO has received lecture fees from Biomerieux, Fresenius Medical and Baxter. CA has received lecture fees from Baxter. LF has received research funding from Baxter and Ortho Clinical Diagnostics, consultancy fees from Medibeacon/La Jolla Pharmaceuticals and honoraria from Biomerieux/Astute.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.