Article Text

Abstract
Objectives After regulatory approval, drug companies, public funding agencies and academic researchers often pursue trials aimed at extending the uses of a new drug by testing it in new non-approved indications. Patient burden and clinical impact of such research are not well understood.
Design and setting We conducted a retrospective cohort study of postapproval clinical trials launched within 5 years after the drug’s first approval, testing anticancer drugs in monotherapy in indications that were first pursued after a drug’s first Food and Drug Administration (FDA) license, for all 12 anticancer drugs approved between 2005 and 2007. FDA, Medline and Embase search date 2019 February 12.
Primary and secondary outcome measures Our primary objective was to measure burden and clinical impact for patients enrolling in these trials. Each trial was sorted into a ‘trajectory’ defined by the drug and cancer indication. The risk was operationalised by proportions of grade 3–4 severe adverse events and deaths. The clinical impact was measured by estimating the proportion of patients participating in trajectories that resulted in FDA approval, uptake into National Comprehensive Cancer Network (NCCN) clinical practice guidelines or advancement to randomised controlled trials within 8 years.
Results Our search captured 104 published trials exploring monotherapy, including 69 unique trajectories. In total, trials in our sample enrolled 4699 patients. Grade 3–4 adverse events were experienced by 19.6% of patients; grade 5 events were experienced by 2.8% of patients. None of the trajectories launched after initial drug approval received FDA approval. Five trajectories were recommended by the NCCN within 8 years of the first trial within that trajectory. Eleven trajectories were advanced to randomised controlled testing.
Conclusions The challenges associated with unlocking new applications for drugs that first received approval from 2005 to 2007 were similar to those for developing new drugs altogether. Our findings can help inform priority setting in research and provide a basis for calibrating expectations when considering enrolment in label-extending trials.
- research ethics
- drug development
- research efficiency
- moral efficiency
- cancer drug development
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Presents a non-economic evaluation of drug development efficiency.
The efficiency of drug development is measured both at the level of drug-indication trajectory and using estimates of patient-level risk and benefit.
The cohort under study was selected to ensure sufficient follow-up time for Food and Drug Administration approval or guideline uptake to occur.
The cohort under study was launched at a time when trial registration was not mandatory, so we rely on clinical trials publications.
Introduction
After new cancer drugs receive regulatory approval, researchers often pursue trials testing the drug in indications or combinations that would extend the use of the drug beyond the indication for which it was initially approved. Sometimes, such efforts are aimed at obtaining a revision to an Food and Drug Administration (FDA) label, other times they aim at discovering an off-label use that can be recommended in clinical practice guidelines.1 Such ‘label-extending’ trials are facilitated by the fact that newly approved cancer drugs are less likely to elicit unexpected safety issues, regulatory approval standards are weaker for postapproval trials,2 and the ease of testing an already approved and manufactured drug are likely to be much less as compared with a drug that has not yet received approval.1 3 4
In numerous cases, label-extending trials have uncovered new uses for approved drugs—especially in the setting of rare malignancies. For example, imatinib was approved for hypereosinophilic syndrome, chronic eosinophilic leukaemia and dermatofibrosarcoma, indications that were not launched into clinical trials until after the initial approval of imatinib in chronic myeloid leukaemia.5 However, where much is known about the volume and clinical impact of prelicense clinical trials in cancer,6 there is much less systematic evidence about label-extending research activities. A previous study suggested that label-extending trials can sometimes be very extensive and failure-prone.7 A 2016 report found that 550 trials of the approved checkpoint inhibitor drugs pembrolizumab and nivolumab were open.8 Such label-extending clinical trial activities, if they do not lead to regulatory approvals or major advances, can deprive other potentially productive lines of cancer drug development of eligible patients and resources. It can also lead to excess patient burden.
We previously reported that combination anticancer therapy drug development efforts launched after a new drug approval are associated with high levels of patient burden but limited clinical impact.9 Such findings can potentially encourage more stringent review when considering proposed postapproval trials for funding or ethical approval. Here we extend those findings to monotherapy anticancer drugs. In what follows, we estimate the patient burden, magnitude of investment and success associated with label-extending monotherapy research efforts for drugs that were newly approved in the years 2005–2007, inclusive.
Methods
Our primary objective was to measure the burden and clinical impact associated with participating in monotherapy trials aimed at extending the label of the 12 novel anticancer drugs that received first FDA approval between 2005 and 2007 inclusive. We included all newly approved drugs within our time frame; drugs that received only a label revision for a new formulation, such as nab-paclitaxel, were excluded. This time frame allowed us to capture publications for all trials launched within 5 years after first approval, affording at least an additional 8 years to account for publication delay after trial closure, uptake in National Comprehensive Cancer Network (NCCN) guidelines or FDA approval. All trials were considered to be part of a ‘trajectory’, which we defined by the drug under study and the indication in which it is being tested (eg, all trials, regardless of the identity of the investigators, testing temsirolimus against prostate cancer would be part of the same trajectory). Our analysis included only trials involving trajectories that were not listed on an FDA label at the time of trial launch.
We operationalised burden with two indicators: the rates of grade 3 or above drug-related adverse events in a single Common Terminology Criteria for Adverse Events (CTCAE) criterion, and the rates of grade 5 drug-related severe adverse events (SAEs).10 We used patient enrolment and number of trials as a proxy for the magnitude of research investment. We used the advancement of a trajectory to FDA approval and recommendation in the NCCN guidelines within 8 years of the launch of a trajectory as proxies for clinical impact; we also measured the fraction of trajectories leading to a randomised controlled trial (RCT). We intentionally chose a sensitive definition of the clinical impact that we and other research groups have used.9 11
Literature search
We searched Medline and Embase (most recent search date 2019 February 12; see the online supplementary file 1) and manually screened for trials of each drug. Our inclusion criteria were: (1) primary data; (2) full text; (3) English language; (4) final report; (5) prospective and interventional trials; (6) administered a drug in our sample in monotherapy in (7) patients with cancer (8) with an enrolment date within 5 years of the drug’s first FDA approval. We excluded studies that were (1) laboratory studies only; (2) case reports; (3) expansion cohorts of published trials; (4) expanded access; (5) mixed malignancy and 6) FDA-approved on-label studies where enrolment began after regulatory approval for the same indication. Adjuvant, maintenance therapy or palliative therapy trials were excluded to focus on monotherapy and curative research efforts.
Supplemental material
Once trials were classified into trajectories, we classified them by whether there was a registry entry in clinicaltrials.gov for at least one randomised clinical trial with a survival endpoint (progression-free survival or overall survival) testing that trajectory (drug and indication) with start dates within 8 years of the trajectory's launch.
Extraction
Extractions were completed according to a previously published template,12 using Numbat Meta-Analysis Extraction Manager.13 Our extraction form (see the online supplementary file 1) captured (a) drug under study; (b) study indication; (c) study characteristics (eg, author-reported phase number, sponsor); (d) trial demographics (eg, number enrolled); (e) enrolment date; (d) primary outcome; (e) treatment-related SAEs.
Trajectories were defined by the anticancer drug being studied and the broad cancer indication explored. Solid tumour indications were classified by anatomical site. In consultation with a haematologist–oncologist after extraction but before analysis, we grouped haematological malignancies into five indication categories (see the online supplementary file 1).
Because the goal of this project was to quantify the burdens and costs expended in label-extending research, the total patient-subject n per trial was extracted as the total number of patients enrolled, regardless of whether they were enrolled on an arm that received the drug in question or were ultimately included in the final safety or efficacy analysis. Trials were marked as having a ‘positive’ result if they met their prespecified primary efficacy outcome with statistical significance and toxicity was reported as acceptable, according to the authors of the publication. SAEs were counted conservatively, capturing only the reported CTCAE10 category with the highest number of adverse events.
Patient and public involvement
No patients were involved in this study.
Analysis and statistics
Analysis of results was conducted using R V.3.4.1.14 Graphics were prepared using the ggplot2 package.15 Pooled SAE rates were calculated using the random-effects inverse-variance meta-analysis method for proportions from the meta package, V.4.8–2.16 Unless explicitly stated otherwise, ranges given after pooled proportions or HRs represent 95% CIs.
Results
Volume and characteristics of label-extending trials testing monotherapy
Our search captured 12 novel anticancer drugs, of which there were 104 label-extending trials of monotherapy, with 69 drug-indication trajectories that were launched within 5 years of the drug’s first FDA approval. The mean number of patients per trial is 45 and the median number of patients per trial is 34.
One or more label-extending trials were pursued for 11 of 12 drugs in our cohort (ranging from 1 to 25 trajectories per drug, see table 1 and Dryad data repository for details).17 Label-extending trials were predominantly single-arm studies (k=99; 95%), phase II (k=96; 92%), followed by phase I trials (k=7; 6.7%) and phase III (k=1; 1.0%). Industry funded 39 trials (38%) in our sample and 57 trials (55%) had non-industry sponsors (the remaining trials did not report a funding source). In 18 trajectories (26%), the first trial was sponsored by industry, whereas there was a non-industry sponsor for the first trial of 45 trajectories (65%; the remaining 6 trajectories had first trials that did not report a funding source). Label-extending trials enrolled 4699 patients.
Sample demographics per drug
Patient risk and burden
Toxicity was reported as acceptable by the authors of the report in 71 trials (68%) in our sample. Among the trajectories represented by our sample, at least 875 patients, 19.6% (17–22%), experienced a treatment-related grade 3–4 SAE, and 59 patients, 2.8% (2.3–3.4%), experienced a treatment-related grade 5 SAE (see Dryad data repository for per trial details).
Advancement to clinical practice, FDA labels and randomised trials
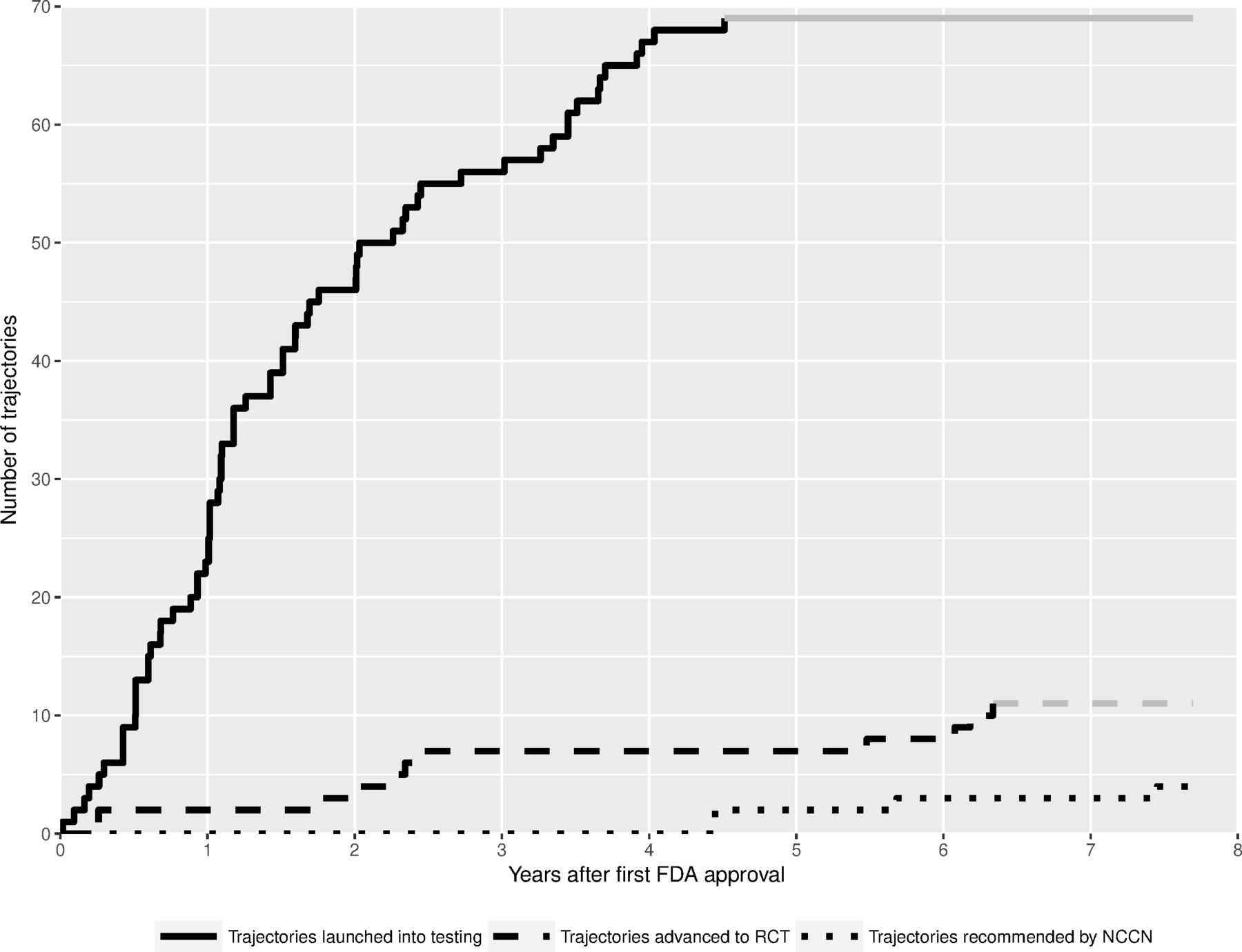
Figure 1 shows the relationship between the launch of new label-extending monotherapy trajectories, their testing in randomised trial, and uptake into NCCN clinical practice guidelines and/or FDA approval.
{kind=link}
Cumulative plot of the number of drug-indication trajectories launched into testing within 5 years of initial FDA approval, trajectories recommended in NCCN clinical practice guidelines within 8 years of trajectory Launch, and trajectories advanced to RCTs within 8 years of trajectory Launch. There were no new FDA approvals in trajectories launched after the initial FDA approval. Grey lines reflect the last value of the specified follow-up period carried forward. FDA, Food and Drug Administration; NCCN, National Comprehensive Cancer Network; RCT, randomised controlled trial.
Label-extending trials with an efficacy endpoint were positive in 37 instances (36%). Eleven trajectories (1.4%) progressed to randomised testing using a survival primary endpoint within 8 years of the first trial in the trajectory. Of the three that had posted results to clinicaltrials.gov as of this writing, one was terminated for insufficient accrual,18 one was terminated early due to SAEs19 and one had a progression-free survival HR that did not show a significant difference between arms.20 There were 24 trajectories in our sample in which more than half of the trials had a positive primary endpoint. Among these trajectories, only four (16%) led to a confirmatory RCT with a survival primary endpoint registered within 8 years of the launch of the trajectory.
No monotherapy trajectories led to a new FDA license within 8 years of the launch of the trajectory. Five trajectories (7.2%) involving two drugs (sunitinib and sorafenib) led to recommendations in NCCN clinical practice guidelines within 8 years of the trajectory’s first clinical trial. One of the new NCCN recommendations was based on an RCT registered within 8 years of its launch (sunitinib for pancreatic adenocarcinoma).21 However, the overall survival endpoint of the trial cited in the NCCN guideline was not statistically significant. See figure 1 for a plot of the dynamics of launching of new trajectories as compared with advancement to RCT and take-up by NCCN clinical practice guidelines.
Discussion
In our cohort of 12 newly approved drugs, 69 label-extending monotherapy trajectories were launched within 5 years of FDA approval. Of these, five (7.2%) led to recommendations in clinical practice guidelines and none resulted in new FDA approvals within 8 years of trajectory launch. Eleven (16%) advanced to randomised phase III trials. These activities involved 104 trials and enrolled 4699 patients and resulted in 875 patients (19.6%) experiencing drug-related adverse events that were grade 3 or 4. A parallel study of postapproval combination therapy exploration in the same set of anticancer drugs found even greater investment, 323 trials of 266 unique combinations, enrolling 29 835 patients. These efforts led to no new FDA approvals of postapproval trajectories launched within five years, and 4.9% of new combinations were recommended in NCCN clinical practice guidelines within 5 years of follow-up.9
Postapproval trials may be more likely than pre-approval trials to target more rare cancer indications and this may affect clinical success rates. Nevertheless, to put our estimates in perspective, the probability that new cancer treatment will advance from entry into phase I testing to FDA approval has been reported to range from 5% to 6.7%.6 22 The frequency with which unapproved cancer drugs in phase II advance to phase III testing was estimated to be 28.3%. Estimates of drug-related grade 3 or 4 adverse events in phase I studies have been reported to be 10.3%.23 For the label-extending trial activities we studied, rates of clinical advancement were lower than estimates for preapproval research, whereas toxicities were higher than those that have been reported for phase I trials.
Our findings have potential implications for human protections, research policy and clinical decision making. The moral basis for trials rests on burdens being redeemed by benefits to volunteers (if any) and new knowledge regarding potential uses of a drug.24 Our analysis provides a baseline for considering the clinical value and patient burden associated with attempted repurposing of new cancer drugs for other malignancies. Such data can be used to explain to patients considering label-extending trials historic rates with which patient contributions have led to major changes in clinical practice.
Our study also has implications for clinical decision making. The volume of label-extending research is such that false-positive signals of treatment activity due to chance are inevitable for some drug-indication trajectories. On the assumption that the null hypothesis in one efficacy trial in each of the trajectories in our sample was true, that statistical tests were two sided, that trial outcomes were independent and that researchers prespecified an alpha of 0.05, the probability that there would be at least one false positive among them is >96%.25 In our sample, five trajectories were recommended for off-label use by the NCCN for the treatment of various malignancies. Given that only one of these trajectories has advanced to an RCT, clinicians writing off-label prescriptions should be aware that some of the evidence on which such recommendations are based may rest on false positives and/or overestimates of effect.
Our findings might also inform research policy. Others have reported high levels of postapproval clinical testing and raised concerns about redundancy and the lack of coordination of postapproval trials.26 One study suggested a relationship between high revenue generation for a drug and postapproval testing.27 Our own team has raised concerns about the use of small postapproval trials creating the suggestion of treatment efficacy (‘clinical agnosticism’) and promoting off-label treatments of approved drugs.28 Poorly justified postapproval trials compete with well-justified trials for a limited pool of study volunteers and research personnel. Research funders and policy-makers might consider incentives that would encourage more selective conduct of postapproval trials and greater efforts towards confirming promising findings from exploratory postapproval studies. Regulators might also reconsider policies that grant companies safe harbour from off-label promotion29 when they circulate reprints of clinical trials testing off-label treatments.26 27 There is high variability in the number of trials pursued per trajectory and also in the number of patients enrolled per trajectory. This confirms a result shown in other work conducted on postapproval trials in the same time period.26 This variability may arise from many different sources: lack of coordination, market considerations, regulatory incentives for rare diseases or individual agendas of small research centres. Our study is not statistically powered to formally test whether factors like market size correlate with research activity or successful research efforts. Previous work suggests relationships between commercial success and medical relevancy may be potential predictors of postapproval trial activity.27
Our analysis has several limitations. First, postapproval clinical trial activity has a low rate of publication,30 and we considered only published monotherapy trials launched within 5 years of the first license. However, given the tendency to selectively report positive trials, we suspect our findings would be even less favourable for label-extending research had we been able to capture the results of unpublished studies. Second, we considered only anticancer drug development; anticancer postapproval clinical testing may be more extensive than other disease areas.26 Third, a more recent sample of drugs—one that includes immunotherapies or precision-medicine drugs—might show greater rates of successful translation. However, all but one of our 12 drugs are targeted agents, and 2 are precision medicine drugs. At the time of the trials in our sample, molecularly targeted agents were taken to be fundamentally different from previous generations of oncology drugs, and there was an expectation that they would lead to lower rates of attrition in drug development.31 We note that clinicaltrials.gov lists a very large volume of label-extending trials for newer drugs. For example, for pembrolizumab, there are at least 30 active label-extending trajectories (search date 2018 October). Fourth, we intentionally chose a sensitive definition of clinical impact. Advancement of a treatment regimen to randomised testing may not, in some cases, represent clinical impact. Last, our analysis is not equipped to morally evaluate the relationship between risk and benefit in label-extending trials. One successful label-extending trajectory—if it entailed a substantial impact on disease—could justify the efforts associated with label-extending research. Many label-extending trials involve rare malignancies.
Label-extending trials offer opportunities to build on safety data accumulated in prelicense research, as well as emerging mechanistic knowledge about a new drug. Despite this, our findings suggest that the challenges associated with unlocking new applications for drugs that first received approval from 2005 to 2007 were similar to those for developing new drugs altogether. Our findings can help inform priority setting in research and provide patients and physicians a basis for calibrating expectations when considering enrolment in label-extending trials.
Acknowledgments
The authors gratefully acknowledge the contributions of Harry Atkins, Gina Freeman, Spencer Hey, Amanda Hakala, Tiger Zheng, Taiji Wang, James Mattina, Yasmina Hachem, Nadine Demko, Natalie McKinnon, Sylviya Ganeshamoorthy, Holly Sarvas and the other members of the STREAM research group.
Footnotes
Contributors BGC and JK: designed this study; wrote the manuscript. BGC: collected data, performed analyses and produced the figure. AD: reviewed and edited the manuscript. All authors read and approved the final manuscript.
Funding This work was funded by CIHR (PJT-148726).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Extra data can be accessed via the Dryad data repository at http://datadryad.org/ with the doi: 10.5061/dryad.zw3r22851.