Article Text

Abstract
Introduction The question of how to administer adequate chemotherapy to synchronise stereotactic body radiation therapy (SBRT) treatment strategy to maximise the benefits of neoadjuvant therapy for the improved prognosis of patients with borderline resectable (BRPC) and locally advanced (LAPC) pancreatic cancer is a challenging and debatable issue. No studies have yet evaluated the efficacy of split-course SBRT as the neoadjuvant chemoradiotherapy regimen. We aimed to study whether neoadjuvant chemotherapy plus split-course SBRT results in better outcomes in BRPC and LAPC patients.
Methods and analysis Treatment-naïve patients with radiographically confirmed BRPC or LAPC, supporting biopsy results and no severe comorbidities will be enrolled. They will be treated with nab-paclitaxel plus gemcitabine (nab-P+Gem) chemotherapy plus split-course SBRT, followed by an investigator’s choice of continuation of treatment with nab-P+Gem or surgery. nab-P+Gem chemotherapy will commence on day 1 for each of six cycles: nab-paclitaxel 125 mg/m2 intravenous infusion over approximately 30–45 min, followed by gemcitabine 1000 mg/m2 intravenous infusion over about 30 min on days 1 and 15 of each 28-day cycle. During the first and second cycles of chemotherapy, SBRT will be given as a single irradiation of 10 Gy four times (days 2 and 16 of each 28-day cycle). The primary endpoint is progression-free survival; while the secondary outcomes are the time to treatment failure, disease control rate, overall response rate, overall survival, R0 resection rate and incidence of adverse effects.
Ethics and dissemination The study protocol was approved by the Ethics Committee of Xiehe Affiliated Hospital of Fujian Medical University (No. 2019YF015-01). Results from our study will be disseminated in international peer-reviewed journals. All study procedures were developed in order to assure data protection and confidentiality.
Trial registration number NCT04289792.
- radiation oncology
- pancreatic disease
- chemotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first study that split-course radiotherapy combined with adequate chemotherapy as neoadjuvant therapy.
Unlike other studies, stereotactic body radiation therapy and chemotherapy are administered as concurrent therapy for patients with borderline resectable pancreatic cancer and locally advanced pancreatic cancer.
A modified regimen of biweekly nab-Paclitaxel plus gemcitabine chemotherapy associated with a lower cost, acceptable toxicity profile is used in this study.
This phase II study design with a relatively small sample size does not allow for subgroup outcomes analysis.
This is a single-arm study without comparison groups or randomisation design.
Background
Value of neoadjuvant therapy in borderline resectable pancreatic cancer and locally advanced pancreatic cancer patients
Pancreatic cancer (PC) is a common malignancy of the digestive system, accounting for about 1%–4% of all malignant tumours.1 It is the fourth-leading cause of cancer deaths in the United States1 and the sixth-leading cause of cancer deaths in China.2 Of the current treatment modalities, surgical resection appears to be the only potential curable option for PC. Unfortunately, owing to its insidious onset, about 80% of patients with PC present with unresectable tumours at initial diagnosis resulting in poor prognosis.3 Moreover, the 5-year survival rate of resectable PC (RPC) after the maximum surgical resection and postoperative adjuvant treatment is only 20%.4 5 To further overcome the shortcoming of traditional adjuvant treatment and improve the curative effect, neoadjuvant therapy has become a research hotspot. The main advantages of neoadjuvant therapy are as follows: (1) It can assess the effectiveness of chemotherapy in advance and screen out patients with very poor prognosis, avoiding meaningless surgery; (2) It can control tumour progression and even reduce the tumour burden to improve the rate of radical operation and (3) It can revert a locally advanced disease into borderline resectable or even resectable status, affording the patient a chance to undergo beneficial surgery.
Although there is a general lack of high-level evidence-based medicine in this regard, most authoritative guidelines and consensus recommend that all borderline RPC (BRPCs) should receive neoadjuvant therapy treatment. At the same time, for locally advanced PC (LAPC) patients, an increasing number of studies have recently reported the possibility of neoadjuvant tumour resection after induction chemotherapy in patients with junctional RPC and unresectable LAPC. This knowledge could further improve the prognosis. Michelakos et al6 analysed the data of 141 BRPC/LAPC patients who underwent surgical exploration after FOLFIRINOX chemotherapy and found that 78% of patients could be successfully resected. Compared with direct surgery, neoadjuvant therapy showed significantly improved disease-free survival (DFS) and overall survival (OS) (DFS: 13.7 vs 29.1 months, p<0.001; OS: 25.1 vs 37.7 months from diagnosis, p=0.01). These results proved that preoperative neoadjuvant therapy is necessary for BRPC and LAPC patients. At present, the indications for neoadjuvant therapy for PC have been expanded. Its safety and effectiveness are being increasingly affirmed by clinical studies. The key to choosing a reasonable neoadjuvant treatment plan is to ensure the efficacy and safety of the therapy to maximise patient benefit.
Choice of neoadjuvant therapy strategy
For patients with potentially RPC and LAPC, chemotherapy-based synchronous radiotherapy or sequential radiotherapy is a widely used clinical solution. At present, although there is no conclusion on whether chemotherapy is needed for combined radiotherapy, this application has been noted in some previous studies.7–11 Accordingly, chemotherapy combined with radiotherapy can significantly improve the surgical resection rate compared with chemotherapy alone (28% vs 12%).12 Therefore, for patients who can tolerate higher-intensity treatment, the combination of radiotherapy may have more enormous survival benefits. Compared with conventional fraction regimen (CFR), stereotactic body radiation therapy (SBRT) has the advantages of short treatment time, high local dose and accurate target location. In a retrospective analysis based on national databases, the OS was significantly higher in PC patients treated with SBRT than CFR.8 Meanwhile, SBRT has shown good results in improving the rate of decline, R0 resection rate and OS as a means of neoadjuvant therapy not only in retrospective cohort studies11 13 but also in prospective trials.14–16
Compared with sequential chemoradiotherapy, concurrent chemoradiotherapy can effectively control the progress of cancer tissues both locally and systemically, and shorten the preoperative treatment time. Although adequate chemotherapy plays an important role in decreasing the rate of metastatic disease and improving long-term survival, the toxic and side effects caused by concurrent chemoradiotherapy (especially with full-dose chemotherapy) in actual clinical practice are too severe to be tolerated. Some patients end up compromising on the dose of chemotherapy and thus do not benefit from adequate chemotherapy for significant treatment outcomes. When it comes to high-dose irradiation in abdominal and pelvic tumours, split-course radiotherapy is a strategy that features an extended interval between fractions instead of daily irradiation to further reducing the toxicity of radiotherapy and improving the patient’s treatment tolerance.17 Moreover, the radiation toxicity can be further reduced by adaptive radiotherapy, which can also increase local control and long-term survival by dose escalation.18–22 Split-course radiotherapy has also played an important role in conventional radiotherapy. As one of the strategies to reduce toxicity in conventional radiotherapy, especially among high-risk patients with locally advanced head and neck cancer and stage III non-small-cell lung cancer patients who are unable to tolerate continuous-course definitive radiotherapy, split-course radiotherapy combined with adequate chemotherapy was shown to be a well-tolerated and effective treatment strategy.23
Although FOLFIRINOX and nab-paclitaxel plus gemcitabine (nab-P+Gem) are both first-line chemotherapy regimens, nab-P+Gem has better progression-free survival (PFS) and tolerance than FOLFIRINOX.24 25 Therefore, in this study, we proposed a comprehensive treatment strategy combining nab-P+Gem chemotherapy (day 1 and 15, 4-week regimen ×6 cycles) with split-course SBRT (10 Gy×4, days 2 and 16, 4-week regimen ×2 cycles).
Methods/design
Study design
This is designed as a single-centre, single-cohort, single-arm, prospective phase II trial in subjects with BRPC or LAPC treated with nab-P+Gem plus split-course SBRT, followed by an investigator’s choice of continuation of treatment with nab-P+Gem or surgery (figure 1).

Overall study design. CR, complete response; CT, computed tomography; IV, intravenous; MRI, magnetic resonance imaging; PD, progressive disease; PET, positron emission tomography; PR, partial response; SD, stable disease.
Primary objective
To evaluate the overall PFS in subjects treated with nab-P+Gem plus split-course SBRT as neoadjuvant therapy, followed by investigator’s choice of treatment. PFS is defined as the time after the first dose of study therapy to disease progression or death (by any cause).
Secondary objectives
To evaluate the time to treatment failure, measured as the time after the first dose of study therapy to treatment failure. Treatment failure is defined as discontinuation of study therapy due to disease progression, death (by any cause), or the start of a non-protocol-defined anticancer therapy.
To evaluate the disease control rate after neoadjuvant therapy, defined as the combined incidence of complete response (CR), partial response (PR) and stable disease (SD) measured at the End of Treatment visit.
To evaluate the overall response rate, defined as the combined incidence of CR and PR.
To evaluate the OS, defined as the time after the first dose of study therapy to death (by any cause).
To evaluate the treatment-related adverse events.
To assess the resection rate and extent of surgical resection (R0 vs R1).
Exploratory objectives
Molecular studies will be conducted to investigate DNA damage, vascular injury, and immune cell infiltration in tumour tissue. In addition, characterisation of early histological and molecular changes, in terms of gene expression, seen within the tumour following treatment will be assessed.
Study population
Treatment-naïve subjects with BRPC or LAPC will be identified as potential study candidates. Among these patients, those that meet the inclusion and exclusion criteria (table 1) will be eligible for final enrolment in the study.
Inclusion and exclusion criteria
Length of study
Enrolment in the study is expected to take about 2 years. The total duration of this study with treatment and follow-up for OS is estimated to last approximately 2 years or until up to 95% of survival data have been collected, whichever comes first.
The End of Trial is defined as either the date of the last visit of the last subject to complete the study, or the date of receipt of the last data point from the last subject required for analysis, as pre-specified in the protocol and/or the statistical analysis plan, whichever is the later date.
Intervention nab-P+Gem
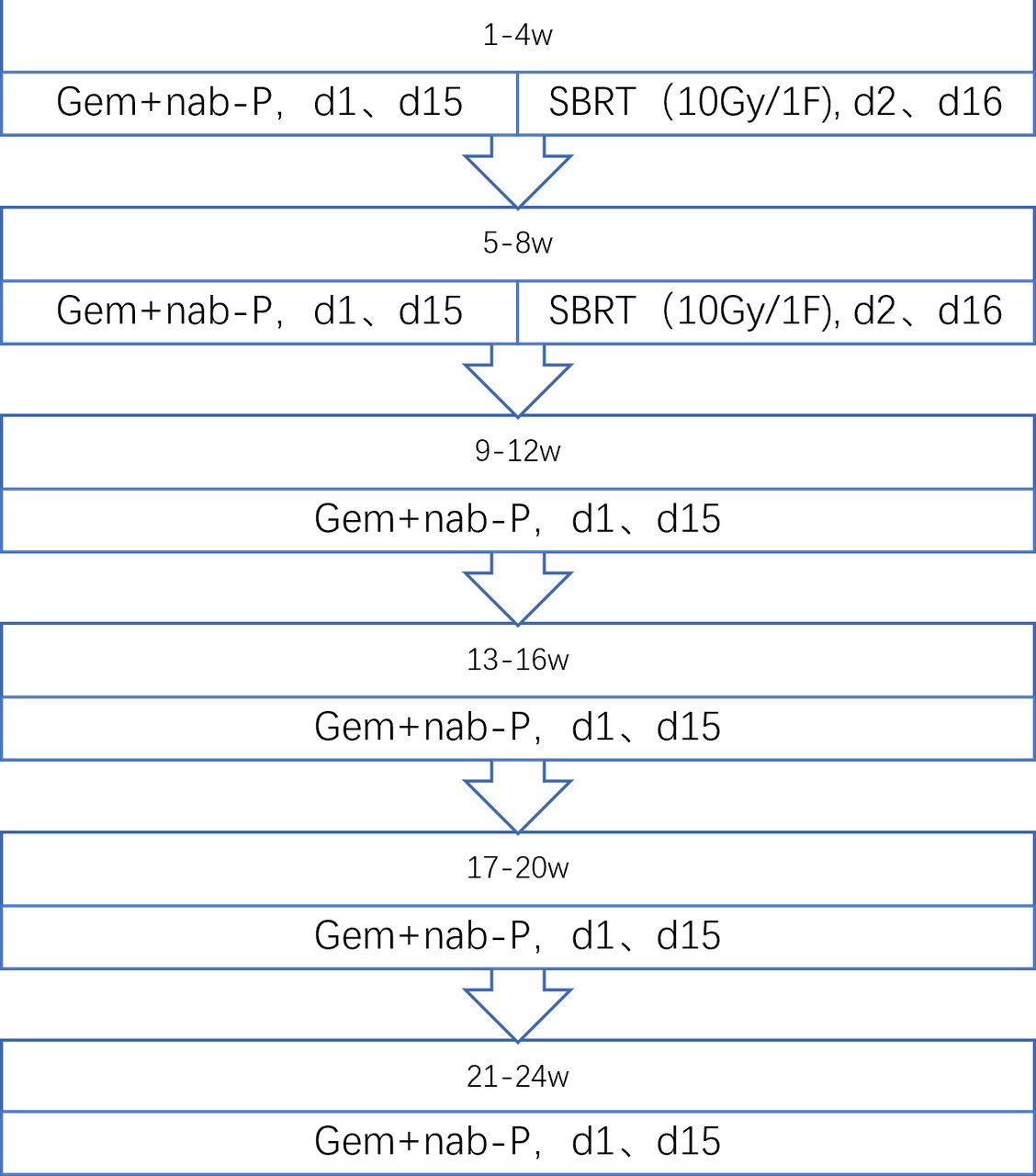
Treatment will commence on day 1 for each of six cycles: nab-Paclitaxel 125 mg/m2 intravenous infusion over approximately 30–45 min, followed by gemcitabine 1000 mg/m2 intravenous infusion over about 30 min on days 1 and 15 of each 28 day cycle (figure 2).
{kind=link}
{kind=link}
Neoadjuvant chemoradiotherapy design. nab-P+Gem, nab-paclitaxel plus gemcitabine; SBRT, stereotactic body radiation therapy.
Due to haematological and other toxicities, the dose may need to be reduced. Dose adjustment will be based on the system showing the greatest toxicity. Toxicity will be graded using the National Cancer Institute’s Common Terminology for Adverse Events (NCI CTCAE) V.5.0. Up to two dose reductions to 100 mg/m2 and 75 mg/m2 for nab-paclitaxel and 800 mg/m2 and 600 mg/m2 for gemcitabine are allowed. If a toxicity requiring dose modification occurs following the second dose reduction of either investigational product, further treatment will be discontinued. In this study, subjects who experience delayed nab-paclitaxel or gemcitabine administration by ≥28 days due to toxicity of the chemotherapy will discontinue further treatment.
Once six cycles have been completed, subjects showing acceptable toxicity levels and those without disease progression will continue on to the investigator’s choice part of the study.
At any time during the study, subjects showing disease progression or unacceptable toxicity levels will be discontinued from the study treatment.
Split-course SBRT
During the first and second cycles of chemotherapy, SBRT will be given as a single irradiation of 10 Gy four times (days 2 and 16 of each 28-day cycle) (figure 2). The SBRT technique in PC has been well described in many published literatures.13 14 16
Patients will be required to be fasting for 4–6 hours before CT simulation. Vacuum bags will be used for immobilisation during SBRT. A plain CT and an enhanced pancreatic parenchymal CT will be performed for radiation treatment planning and target delineations. The positioning will be performed by a large-aperture multirow spiral CT simulator (Philips Medical Systems, Highland Heights, Ohio, USA). The scanning range will be from T4/T5–L5/S1 in the exhalation position, including the whole pancreas, at least 10 cm above and below the tumour, with a slice thickness of 1.25 mm.
SBRT will be delivered via CyberKnife (Accuray Incorporated, Sunnyvale, California, USA), an image-guided frameless stereotactic robotic radiosurgery system. All patients will be implanted with gold fiducials guided by endoscopic ultrasound within or adjacent to the tumour. The gross tumour volume (GTV) is delineated as a radiographically evident gross disease by enhanced CT. Clinical target volume (CTV) encompassing areas of the potential subclinical disease spread is also designated at the discretion of the physician. In most cases, the CTV equals to GTV. Planning target volume (PTV) includes a 2–5 mm expansion margin on CTV. When the tumour is close to critical organs, the margin expansion is allowed to be nonuniform26; the expanded PTV outside of CTV should be avoided in this direction. At least 90% of PTV should be covered by the prescription dose. Normal tissue dose constraints are referred to the American Association of Physicists in Medicine guidelines in Task Group-101.27
According to NCI CTCAE V.5.0, radiotherapy does not need to be adjusted when grade 3 toxicity unrelated to radiotherapy occurs. However, if the toxicity develops to grade 4, radiotherapy should be stopped until toxicity is controlled. If grade 3 radiotherapy-related acute toxicity occurs during treatment, the radiotherapy dose is only withheld if symptoms cannot be relieved after treatment. Up to two dose reductions to 8 Gy and 6 Gy are allowed. If a toxicity requiring dose modification occurs following the second dose reduction of irradiation, further treatment should be discontinued. Subjects who experience delayed irradiation by ≥28 days owing to radiotherapy-related acute toxicity would discontinue further treatment. If there is a grade 4 radiotherapy-related toxicity during treatment, subjects will be discontinued from the radiation treatment
Investigator’s choice
For subjects who complete 6 cycles of nab-P+Gem without disease progression or unacceptable toxicities will undergo surgical exploration, the investigator will determine which of the following options are best for the subject: (1) continuation of nab-paclitaxel and gemcitabine therapy until disease progression or unacceptable toxicity.
(2) Surgical intervention: If the tumour response allows for surgical intervention, the subject will be eligible for surgery that the investigator deems appropriate. If subjects demonstrate a major response to therapy, surgical intervention may occur prior to completion of the planned six cycles of nab-P+Gem.
Follow-up
Subjects who discontinue treatment with nab-paclitaxel and gemcitabine, chemoradiation or surgery for any reason will have a safety follow-up visit 28 days after treatment discontinuation and will be followed up for disease progression approximately every 56 days. All subjects will be followed up for OS and poststudy anticancer therapies approximately every 90 days by phone or review of medical records until death, withdrawal of consent, or lost to follow-up. At any time during the study, subjects with disease progression or unacceptable toxicity will be discontinued from the study treatment. During the study, subjects, including those who discontinue treatment without disease progression, will have CT/MRI scans every 56 days (−3/+7 days) until documented progression of disease, withdrawal of consent from active participation in the study, lost to follow-up or death, whichever is earliest. Tumour evaluations will be assessed by the investigators and responses will be determined according to the RECIST V.1.1 guidelines.
Sample size and statistical analysis
The primary endpoint for this study is the median PFS. The null (Ho) and alternative (Ha) hypotheses for this endpoint are as follows, where M is the median PFS and 6.8 months is the median PFS based on the data of our research centre and the previous research reports25 28–30: Ho: M≤6.8 Ha: M>6.8.
A total sample size of 27 subjects will have 80% power to detect an increase in the median PFS of 6.8–12 months. The sample size is calculated assuming a one-sided alpha of 0.05 that subjects will be enrolling for 24 months, and that each subject will be followed up for a minimum of 1 year. Twenty-seven subjects will be enrolled assuming a 10% drop-out rate.
For continuous variables, statistical normality will be first assessed. Comparisons between these main effects will be then performed by analysis of variance or its two non-parametric equivalents as appropriate. Categorical data will be compared by χ2 tests or Fisher’s exact tests. The OS and PFS curves will be plotted by the Kaplan-Meier method. The log-rank test will be employed for comparisons of OS and PFS. Proportional hazard Cox regression adjusted for variables will be used when necessary. Data analysis will be performed on both intention-to-treat and per-protocol basis. All p values will be reported using a significance level of 0.05.
Patient and public involvement
Patients were not involved in the idea conception of this trial.
Patients were not involved in the design of this study nor in recruitment of the study.
Discussion
Clinically, PC can be divided into four categories according to resectability status: RPC, BRPC, LAPC and metastatic disease. The advantages of neoadjuvant therapy include a lower probability of postoperative recurrence and metastasis, better downstaging and improved rate of tumour R0 resection.24 Based on the above advantages of neoadjuvant therapy, the National Comprehensive Cancer Network (NCCN) guidelines, 2013 edition, started to recommend that RPC and BRPC with high risk factors of recurrence or metastasis should be treated with neoadjuvant therapy. Given the local involvement of adjacent blood vessels, LAPC is regularly considered unresectable; chemotherapy combined with radiotherapy is a conventional treatment option recommended by most guidelines for such patients. Recently, more and more studies have reported the possibility of tumour resection after induction chemoradiotherapy in BRPC and LAPC, which can further improve the prognosis.6 31 32 The NCCN guidelines recommended operative treatment to patients from those who respond to neoadjuvant treatment in vision 2017 to those who have no progression after neoadjuvant treatment in vision 2018. At present, not only the clinical indication of operation but also the application of neoadjuvant therapy for the BRPC and LAPC has been expanded, and the safety and effectiveness of neoadjuvant therapy have been confirmed in clinical practice. It is important to choose a reasonable neoadjuvant therapy scheme to maximise the benefits for patients.
For patients with BRPC and LAPC, concurrent chemoradiotherapy or sequential radiotherapy following chemotherapy is a clinically widely used regimen.33 34 Usually, conventional radiotherapy delivers doses of 45–54 Gy in 1.8–2.0 Gy fractions, which result in a long treatment period and may result in micrometastases during neoadjuvant therapy.35 Because to its delivery of high biologically effective doses within a short period and quick dose fall-off, SBRT may not delay subsequent treatment and provide high pathological CR rates with acceptable toxicities simultaneously, combined with chemotherapy. Hence, SBRT may be a more appropriate choice than conventional radiotherapy as a new adjuvant therapy. Nowadays, SBRT with high-dose irradiation is widely used, and cyber-knife technology can maximally focus the radiation on the tumour to limit radiotoxicity. However, it still cannot completely avoid adjacent organs and tissues from radiation damage, as particularly in PC, the tumour is close to the duodenum and gastrointestinal tract among other digestive organs.36 Although adequate chemotherapy plays an important role in decreasing the rate of metastatic disease and improving long-term survival, therapeutic toxicity of SBRT limits the dose of chemotherapy in concurrent chemoradiotherapy. Split-course radiotherapy is a strategy mainly proposed for the purpose of further reducing the toxicity of radiotherapy and improving the patient’s treatment tolerance, with the added feature of an extended interval between fractions instead of daily irradiation.17 37 38
Although it might be implied from previous studies that split-course radiotherapy with or without chemotherapy appears to be a well-tolerated and effective treatment method, potential benefits from split-course SBRT in neoadjuvant chemoradiotherapy still need to be confirmed in prospective studies. Therefore, our aim herein is to investigate whether adequate nab-P+Gem chemotherapy (days 1 and 15, 4-week regimen ×6 cycles) with split-course SBRT (10 Gy×4, days 2 and 16, 4-week regimen ×2 cycles) as a neoadjuvant treatment strategy can prolong survival for BRPC and LAPC patients with well tolerance. There is no phase I trial of this new chemoradiotherapy combination. However, in our centre, some patients with metastatic PC have adopted this treatment scheme as palliative treatment, which was well tolerated. We will also make a close toxicity follow-up in the first enrolled patients.
The superiority of sequential chemoradiotherapy in controlling local/regional lesions and improving patient survival has been demonstrated in most solid tumours. The rationale for concurrent chemoradiotherapy is that these drugs kill tumour cells by their own cytotoxicity and by increasing the sensitivity of tumour cells to radiation. Paclitaxel and gemcitabine are classic chemotherapeutic agents that sensitise cells to radiotherapy. The cells in the G2/M phase are most sensitive to radiation, while those in the S phase have significant radiation resistance. Paclitaxel impose a significant G2/M block by inhibiting spindle formation in the mitotic phase.39 Gemcitabine can inhibit various enzymes involved in DNA synthesis and repair in the S phase, leading to fewer tumour cells in phase.40 However, the limitation of concurrent chemoradiotherapy is that it increases the toxic side effects and reduces the patient’s tolerance. Split-course SBRT can increase not only the local control of the primary tumour mass (which can often be achieved by high-dose radiation) but also reduce normal-tissue toxicity (which can often be achieved by long treatment intervals).
On the basis of present studies, 2–4 weeks fraction intervals applied in split-course SBRT have ensured either a significant decrease in the GTVs or stable disease without significant repopulation of tumour cells.41–43 Moreover, our study combined split-SBRT with concurrent full-dose chemotherapy that can further reduce the tumour from repopulating. Hypoxia-induced radiation resistance is still a concern in SBRT, and owing to the weakening of SBRT reoxygenation,44 45 hypoxia may become one of the important mechanisms of SBRT radioresistance. Reoxygenation during the 2-week radiation interval in our study may effectively reduce the hypoxia-induced radiotherapy resistance.
In the SBRT treatment mode, in addition to the classical ‘4R,’ researchers believe that there are unique high-dose radiation-induced vascular injury effects different from conventional irradiation that can induce secondary cell death by causing damage to the tumour vasculatures. At the dose of conventional fractionated radiotherapy (1.5–2.0 Gy/F), the effect of radiation on the blood vessels is mild, and the changes can be restored to baseline levels in the later stages of radiotherapy. A single dose of 5 Gy may be the threshold of vascular permeability change and endothelial injury in tumours. Irradiation of human tumour xenografts or rodent tumours with 5–10 Gy in a single dose causes relatively mild vascular damages, but increasing the radiation dose to higher than 10 Gy/fraction induces severe and unrepairable vascular damage resulting in reduced blood perfusion.46 In this trial, 10 Gy has been selected as the fractional dose to bring about repairable vascular effects. At the same time, irradiation will be performed on the second day of each drug treatment, followed by a 2-week interval between irradiation and chemotherapy to allow the recovery of vascular damages, so that the irradiation will not affect the efficacy of chemotherapy.
SBRT-induced immunogenic cell death can lead to a massive release of tumour-associated antigens and various proinflammatory cytokines, thereby triggering a specific antitumour immune response.47 48 However, immune effects after SBRT may vary from different fractioned patterns and interval time.49–52 These data suggest that there may be a threshold dose that effectively activates immunity, and that doses below the threshold may not be effective for immune stimulation, while those above the threshold will produce immunosuppression. In most researches, 10 Gy is considered an effective dose to activate immunity. In continuous high-dose irradiation, the radiation may kill the immune cells around the tumour indiscriminately, leading to reduced antitumour immune effect.53 In this trial, the prolonged interval time may enhance immune effects by decreasing the damage to surrounding recruited immune cells.
Data collection/management/monitoring
Data collection and management
Data collections and evaluations will be according to the timeline shown in tables 2–4. Pretreatment status, efficacy and toxicity of chemotherapy/SBRT and postoperative complications will be evaluated and recorded by the physicians. Furthermore, to promote data accuracy and completeness, all information about the pretreatment assessment and follow-up will be carefully checked by the physicians first and rechecked by the researchers not involved in the study.
Table of events—induction: nab-paclitaxel plus gemcitabine
Table of events—investigator’s choice: continuing with nab-paclitaxel plus gemcitabine
Table of events—investigator’s choice: surgery as per institutional standard of care
After completing the above six cycles of treatment, the investigator will select the best treatment according to the actual clinical situation. Please refer to tables 3 and 4 for details. The study will be withdrawn if the disease progresses or the toxicity becomes intolerable during treatment.
Patients’ pseudonymous medical records and information will be extracted from the database and reviewed for trial purposes, but their data would be kept strictly confidential within the study. Until a valid consent has been obtained, investigators will not assume any demands, including publishing or reporting of individual patient’s data, especially the data required for this clinical trial.
After the completion of the trial, documents of the trial will be maintained for at least 5 years according to the Chinese Good Clinical Practice (GCP)-Regulation. The Research Unit of the Medical Affair Department of the Xiehe Affiliated Hospital of Fujian Medical University (XAHFMU) will be responsible for archiving all relevant data of the trial.
Data safety monitoring board
The Institution Review Board of Fujian Medical University will act as the data safety monitoring board to monitor the recruitment, the report of adverse events and the data quality semiannually.
Ancillary and post-trial care
Patients enrolled into the study are covered by indemnity for negligent harm through the standard National Health Service Indemnity arrangements. Patients will have insurance to cover for non-negligent harm associated with the protocol given by the University of Fujian Medical, including additional healthcare, compensation or damages whether awarded voluntarily by the Sponsor, or by claims pursued through the courts. However, incidences judged to arise from negligence (including those due to major protocol violations) will not be covered by study insurance policies.
Ethics and dissemination
The study protocol was approved by the Ethics Committee of XAHFMU (No. 2019YF015-01). The study was registered at the Chinese Clinical Trial Registry on 6 July 2019 (ChiCTR1900024345). We will fully inform the eligible patients of the purpose and procedures of this study. Written informed consent will be obtained after patients decide to participate. All clinical data are collected by research members confidentially. We will present our findings through scientific publication in international peer reviewed journals as well as at international and national conferences.
Acknowledgments
We appreciate Dr Bangwei Zeng for his precise comments on the revisions of the protocol and article.
References
Footnotes
RZ and CW contributed equally.
Contributors BX and HH conceived and designed the study. RZ and XH drafted the manuscript. RZ and CW and will be in charge of conducting the trial. CW and HH contributed the section pertaining to surgery and will perform the surgery and assess the postoperative situation of patients. CW and DH are responsible for the chemotherapy aspect of the study design and execution. X-BL and QL are responsible for treatment plans of radiotherapy. All the authors approved of the final version for submission.
Funding This study was supported by Joint Funds for the Innovation of Science and Technology, Fujian province (grant number: 2017Y9050).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Patients' consents for publication of data have been obtained.
Provenance and peer review Not commissioned; externally peer reviewed.