Article Text

Abstract
Introduction The time-lapse imaging system (TLS) is a newly developed non-invasive embryo assessment system. Compared with conventional incubators, a TLS provides stable culture conditions and consistent observations of embryo development, thereby potentially improving embryo quality and selection of the best quality embryo. Although TLSs have been routinely used in many in vitro fertilisation (IVF) centres globally, there is insufficient evidence to indicate that TLSs result in higher cumulative live birth rates over conventional incubators. The purpose of this study is to compare the cumulative live birth rates and safety including miscarriage in infertile patients with diminished ovarian reserve (DOR) from both TLSs and conventional incubators.
Methods and analysis This study is a double-blind randomised controlled clinical trial (1:1 treatment ratio of TLSs vs conventional incubator). A total of 730 patients with DOR undergoing the first or second cycle of IVF or intracytoplasmic sperm injection (ICSI) will be enrolled and randomised into two parallel groups. Participants will undergo embryo culture in the TLSs (group A) or the conventional incubators (group B), respectively. Embryos are selected for transfer in both groups by the morphological characteristics. The embryo selection algorithm software is not used in the TLSs. The primary outcome is the cumulative live birth rate of the trial IVF/ICSI cycle within 12 months after randomisation. This study is powered to detect an absolute difference of 10% (35% vs 25%) at the significance level of 0.05% and 80% statistical power based on a two-sided test.
Ethics and dissemination This trial has been approved by the Institutional Ethical Committee of Shanghai First Maternity and Infant Hospital (KS1958). All participants in the trial will provide written informed consent. The study will be conducted according to the principles outlined in the Declaration of Helsinki and its amendments. Results of this study will be disseminated in peer-reviewed scientific journals.
Trial registration number Chinese Clinical Trial Registry (ChiCTR1900027746).
- embryology
- fetal medicine
- reproductive medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first randomised controlled trial with a relatively large sample size comparing the effectiveness of time-lapse imaging systems (TLSs) with conventional incubators in infertile patients with diminished ovarian reserve.
The primary outcome of this study is the cumulative live birth rate, which reflects the ultimate goal of couples undergoing fertility treatment.
This study includes different controlled ovarian stimulation protocol which reflects the variation found in real-world clinical practice.
The sample size calculation is based on a difference of 10% in the cumulative live birth rate between the two groups, thus a smaller difference in the cumulative live birth rate may not be detected.
This study does not assess embryo selection, thus the full benefit of TLSs will not be evaluated.
Introduction
Since the first baby was born in 1978 by in vitro fertilisation-embryo transfer (IVF-ET), IVF has been used worldwide. More than seven million babies have been born following assisted reproductive technologies (ART).1 In the past 40 years, despite continuous efforts in optimising ART procedures, implantation rates of IVF embryos are still low, and the clinical pregnancy rate has not increased significantly in recent years. The latest data from European countries in 2017 and 2018 indicate a clinical pregnancy rate of approximately 35% per transfer cycle.2 3 To achieve better pregnancy outcomes, more studies are needed that ultimately improve embryo culture environment, embryo quality and optimisation of embryo selection with developmental potential for transfer and freezing.
In vitro culture and selection of embryos are vital steps for all ART procedures. At present, reliance on morphological characteristics remains the most common method for assessing embryo developmental potential. Such an assessment is not objective and comprehensive enough to observe embryonic development, thus important developmental events might be missed. The time-lapse imaging system (TLS) is a newly developed non-invasive embryo quality assessment method. Compared with the conventional culture system, TLS provides stable culture conditions, and reveals the details of the early stages of human embryo development. Studies have shown that two morphologically similar embryos may have completely different developmental processes if analysed by a TLS.4 Therefore, TLSs can exclude some abnormal cleavage embryos that are considered to have ‘normal’ developmental potential in static assessment by using conventional incubators. Taken together, TLSs may help embryologists to select embryos with developmental potential for transfer, which can help to improve implantation rates and clinical pregnancy rates in infertile patients.5
Although TLSs have been routinely used in many IVF centres globally and are used as an ‘add-on’ only in a few IVF centres in China with a large number of IVF cycles, it is unclear that if the TLS results in higher live birth rates because high-quality evidence is limited. Retrospective studies and cohort studies have shown that TLSs improved embryo quality and selection, and improved clinical outcomes.6 7 To date, there are only a few randomised controlled trial (RCT) studies assessing the effectiveness of TLSs compared with conventional incubators. Data from recent meta-analyses showed conflicting results comparing TLSs with conventional incubators.8–11 These meta-analyses included studies with different populations. Confounding factors such as culture conditions (culture medium, O2 concentration), day of transfer, the number of embryos transferred, fresh or frozen embryo used, oocyte source, patient population, the TLS type and algorithms to predict various clinical outcomes, were inconsistent among the included studies.12 Moreover, these studies were at high risk of bias for randomisation and allocation concealment, thus results should be interpreted with extreme caution. Notably, most of the studies investigated patients with normal ovarian response or good prognosis, and only one pilot RCT has been conducted on the effectiveness of TLSs in patients with poor prognosis.13
Diminished ovarian reserve (DOR) generally refers to a quantitative decline of the oocyte pool, shown as an abnormal ovarian reserve test.14 15 There is no internationally agreed definition of DOR. The ESHRE Bologna criteria of DOR are antral follicle count (AFC) <5–7 follicles or anti-Mullerian hormone (AMH) <0.5–1.1 ng/mL,14 whereas the POSEIDON criteria defined DOR as AFC <5 follicles or AMH <1.2 ng/mL.15 Patients with DOR usually showed fewer follicles in the ovarian stimulation cycle, lower blood oestradiol levels, more gonadotropin (Gn) usage, high cancellation rate, low number of oocytes retrieved and low clinical pregnancy rate.14 Management of women with DOR is one of the major challenges in reproductive medicine.
With the delay of childbearing and the implementation of China’s second birth policy, the number of ART cycles in women with DOR has been increased in recent years. Therefore, we propose a randomised controlled clinical trial to compare the cumulative live birth rate of TLSs with conventional incubators in patients with DOR.
Methods and analysis
Study design and setting
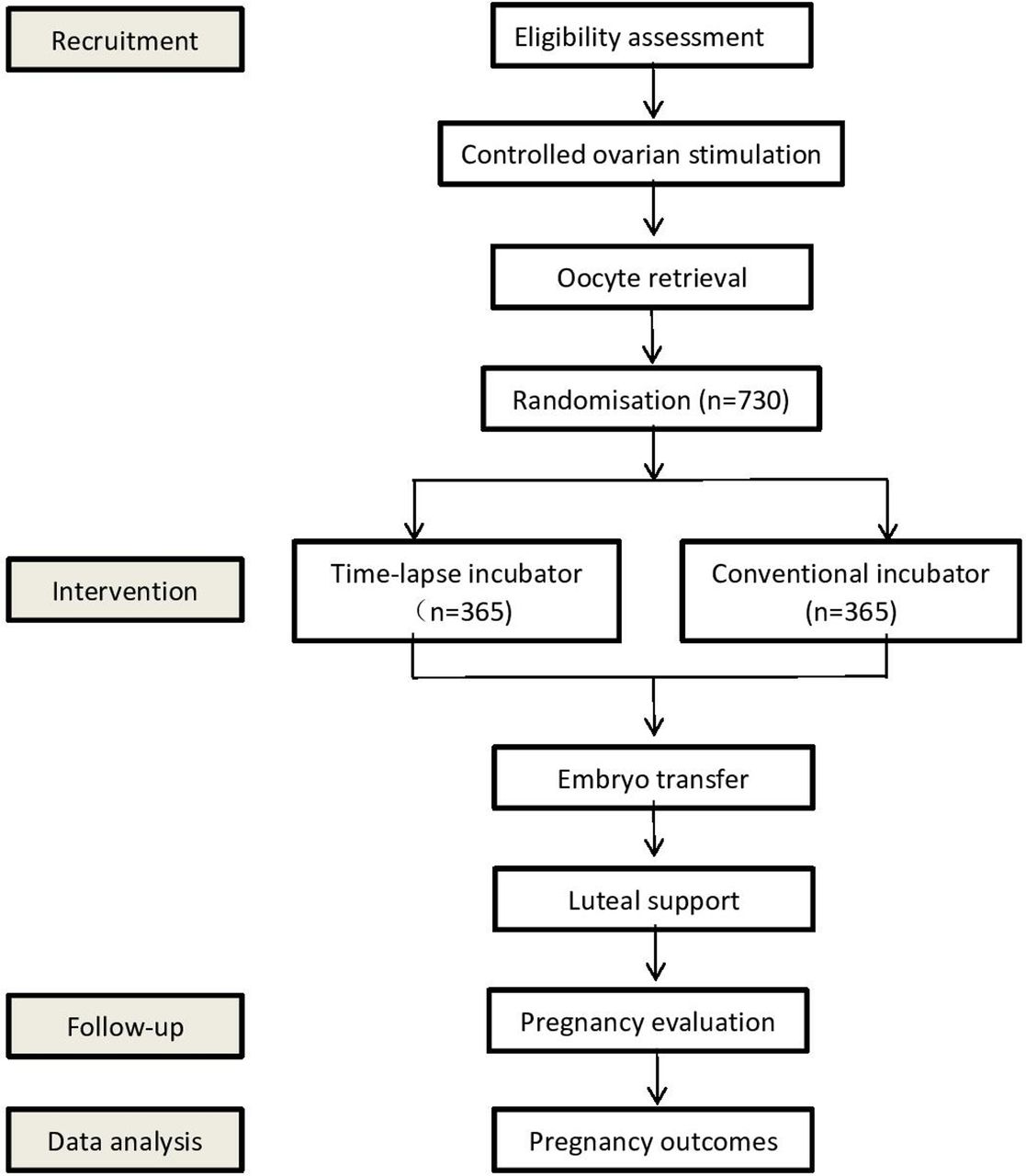
This study is a single-centre, parallel, double-blind, superiority randomised controlled clinical trial (1:1 treatment ratio). Participants will be recruited from Shanghai First Maternity and Infant Hospital. This protocol has been written following the Standard Protocol Items: Recommendations for Interventional Trials. The trial design is summarised in figure 1, whereas the schedule of enrolment, interventions and assessments during the study period is shown in table 1.
Schedule of enrolment, interventions and assessments
{kind=link}
Flowchart followed the checklist of Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) showing patient enrolment, allocation, treatment and follow-up of participants.
Inclusion criteria
Infertile couples scheduled for their first or second IVF/intracytoplasmic sperm injection (ICSI) cycle.
In line with DOR criteria (AFC <7 follicles or AMH <1.2 ng/mL).
Exclusion criteria
Couples undergoing pre-implantation genetic testing.
Women over 45 years of age.
Women with congenital or secondary uterine abnormalities, such as uterine malformations including the single-horned uterus, septate uterus or double uterus, adenomyosis, uterine submucosal fibroids, intrauterine adhesions.
Women with hydrosalpinx.
Women with recurrent miscarriage.
Women with polycystic ovary syndrome.
Women with endocrine or metabolic abnormalities (pituitary, adrenal, pancreas, liver or kidney).
Recruitment
Infertile couples who come to the outpatient clinic to receive IVF/ICSI will be screened by a trained clinical team, familiar with the selection criteria. Eligible patients will then be approached by a member of the research team who will explain the trial details before the commencement of the IVF/ICSI treatment. Couples will be offered time to consider ongoing participation in the trial. Those couples who refuse to participate will be treated according to the conventional protocols at the centre. The decision to refuse or withdraw from the study will not affect their conventional clinical treatments and their relationship with clinical practitioners.
Randomisation and blinding
Eligible women will be randomised to use TLSs or conventional incubators. Randomisation and allocation of patients to study arms will be performed after the oocyte retrieval (on the same day). Permuted block randomisation is formulated by collaborative investigators who are not involved in the consulting and treatment procedures and a computer-generated randomisation list in a 1:1 ratio, with a variable block size of 2, 4 or 6 is stored in an online trial system (REDCap). When there is an eligible participant to be enrolled in the study, an embryologist will log in to the system to get the allocation of the participant.
Except for participating embryologists and laboratory technicians, this study will be blinded to participants, clinicians, investigators, ultrasonographers and nurses who conduct follow-up until the completion of statistical analysis of this study.
Interventions
All participants will receive controlled ovarian hyperstimulation (COH) treatment, which is performed by standard routines at the study centre. The selection of protocols will be completed by physicians, who are blinded for group allocations. In the gonadotropin-releasing hormone antagonist (GnRH-ant) protocol, all participants will receive daily injection Gn (Gonal-F or Puregon or HMG) from day 2 or day 3 of the menstrual cycle. When at least one follicle has reached a diameter of 12 mm or on day 6 of ovarian stimulation, GnRH-ant (Cetrotide or Ganirelix) 0.25 mg/day will be administered subcutaneously until the trigger day (include the trigger day). For the long GnRH-agonist (GnRH-a) protocol, pituitary downregulation will be initiated 7–10 days before the menstrual cycle with GnRH-a (subcutaneous Triptorelin 0.1 mg/day or intramuscular Triptorelin 1.25–1.88 mg one-time). After 10–14 days or on day 2 of the menstrual cycle, Gn treatment will be commenced. For the short GnRH-a protocol, mild stimulation protocol or progestin-primed ovarian stimulation protocol, participants will receive Triptorelin (subcutaneous injection 0.1 mg/day), oral Clomiphene citrate (50–100 mg four times per day) or oral Duphaston (10 mg two times per day), respectively on day 2 or 3 of the menstrual cycle, and Gn will be used at the same time.
For all the protocols, menstrual cycles of patients can include spontaneous menstrual cycle, and irregular menstrual cycle by the use of oral contraceptives (OC) or progestins. Before COH treatment, baseline pelvic ultrasound will be undertaken. As well, baseline serum hormones such as follicle-stimulating hormone, luteinising hormone, oestradiol (E2), progesterone (P) and β-hCG will be measured. The starting dose of Gns is 150–300 IU/day for the first 4 days based on the characteristics of each patient. Transvaginal ultrasound scanning and hormonal measurement will be repeated every 2–3 days to monitor follicle growth. The subsequent dose of Gn will be adjusted according to the individual response. After two or more follicles reach a diameter ≥18 mm, 250 µg of hCG will be once injected on trigger day. In women with hyper-response (≥15 follicles ≥12 mm), 0.2 mg Triptorelin, or 4000 IU of hCG will be administered.
Oocyte retrieval is scheduled for 36 hours (±2) after hCG injection. Fresh ejaculated semen samples will be obtained on the day of oocyte retrieval, and then prepared by swim-up protocol according to routines.16 The IVF procedures at the study centre have been previously described.16 All the oocytes will be inseminated with 2–5×106 per oocyte motile spermatozoa approximately 39–42 hours after hCG injection. Gametes are then co-incubated overnight at 37°C under 5% O2 and 6% CO2 in the conventional incubators. Assessments of fertilisation are carried out approximately 16–18 hours (day 1) after insemination. Then zygotes are left individually in conventional incubators for a further 48 hours or are transferred to the pre-equilibrated EmbryoSlide for culture within the EmbryoScope (Vitrolife) according to the randomisation results. The infertile couples with male factor such as oligoasthenospermia will undergo ICSI as per the routines at the study centre. For couples who take ICSI, the oocytes will be denuded by hyaluronidase before micromanipulation. Only the mature, metaphase-II oocytes with an extruded first polar body are microinjected. The procedure of ICSI has been previously described.17 After injection, oocytes are transferred to the pre-equilibrated EmbryoSlide for culture within the EmbryoScope or standard culture dishes and cultured individually in conventional incubators (OriGen) according to the randomisation results. Assessments of fertilisation and embryo quality after ICSI for the conventional incubators group are identical with IVF. The images taken by the TLS are reviewed at 16–18 hours post-injection for fertilisation assessment. The cleavage embryo quality for both groups will be observed at 48 (day 2) or 72 (day 3) hours after oocyte retrieval. The embryos are scored as grade 1 to grade 6 according to the quality, numbers, size of the blastomeres and the amount of anucleate fragmentation. Briefly, grade 1 embryos had equal-sized blastomeres and no cytoplasmic fragments; grade 2 embryos had equal-sized blastomeres and less than 25% fragmentation; grade 3 embryos had blastomeres of distinctly unequal size and no cytoplasmic fragments; grade 4 embryos had blastomeres of distinctly unequal size and less than 25% fragmentation; grades 5–6 embryos had more than 25% and 50% fragmentation, respectively. Embryos of 6–8 cells and of grade 1 or 2 were defined as good quality embryos in this study.16 Embryos are selected for transfer in both groups based on morphological characteristics. The embryo selection algorithm software is not used in the TLSs.
For participants receiving fresh ET, this transfer will be performed on day 3 after oocyte retrieval under ultrasound guidance. Surplus embryos will be frozen according to the routines at the study centre. Luteal support is administered in the form of vaginal P (Crinone) 90 mg/day until confirmation of biochemical pregnancy, and will be maintained to 10 weeks of gestation. The P will be discontinued if a biochemical pregnancy is not observed. Oral P (20 mg two times per day; Duphaston) will be administered to women who present with vaginal bleeding.
The criteria for elective cryopreservation include increased risk of ovarian hyperstimulation syndrome and elevated P levels in this study. For participants undergoing frozen-thawed ET, patients with irregular menses will receive oral E2 valerate (Progynova) 4–6 mg/day on days 2–3 of a subsequent artificial menstrual cycle (by the use of OC or progestins) within 6 months after oocyte aspiration. Oral P will be added if the endometrial thickness is ≥8 mm. Patients with regular menses will have ovulation monitoring by transvaginal ultrasound from day 12 of the menstrual cycle. Oral P will be added on the day of ovulation. Frozen-thawed embryos will be transferred on day 3 after P initiation. The transfer procedure will be the same as that used for the fresh ET. Oral medications will be continued at an unchanged dose until the confirmation of biochemical pregnancy and will be maintained to 10 weeks of gestation. The P will be discontinued if a biochemical pregnancy is not observed.
Follow-up
Urine and blood hCG will be measured 14 days after ET, with positive results indicative of a biochemical pregnancy. If the gestational sac is observed with ultrasonography on 7 weeks after transfer, clinical pregnancy will be confirmed. Ongoing pregnancy is defined by the presence of a gestational sac with a fetal heartbeat after 12 weeks of gestation.
For women who have an ongoing pregnancy confirmed, they will be required to notify researchers of the time of delivery. In 2 weeks after delivery, the information of pregnancy (pregnancy complications and fetus information), delivery information (gestational age, delivery mode, placenta abnormality and/or delivery complications), infant information (such as sex, birth weight, birth defect) will be collected by completing forms.
Outcome measures
The primary outcome will be the cumulative live birth of the trial IVF/ICSI cycle per woman randomised within 12 months of randomisation. This will not include live birth in subsequent IVF/ICSI cycles if the trial cycle failed as well as live birth that happens after 12 months of randomisation even if it is a result of the trial IVF/ICSI cycle. This will also exclude spontaneous pregnancies because they are not outcomes of the trial IVF/ICSI cycle and not related to TLSs. Live birth will be defined as the delivery of one or more living infants (≥22 week’s gestation or birth weight more than 500 g).
For the effectiveness of the treatment, we will record the following secondary outcomes in terms of effectiveness:
Good quality embryo: defined as embryos with 6–8 cells and ≤25% fragmentation developed from 2PN embryos on day 3 observation.
Biochemical pregnancy: defined as blood hCG ≥10 U/L at 14 days after ET per woman randomised.
Clinical pregnancy: defined as one or more observed gestational sacs or with fetal heartbeat under ultrasonography at 7 weeks after ET per woman randomised (including clinically documented ectopic pregnancy).
Ongoing pregnancy: defined as the presence of a gestational sac and fetal heartbeat after 12 weeks of gestation per woman randomised.
For the safety of the treatment, we will record the following treatment complications as secondary outcomes:
Miscarriage: defined as the spontaneous loss of an intrauterine clinical pregnancy prior to 22 completed weeks of gestational age.
Ectopic pregnancy: defined as the implantation that takes place outside of the uterine cavity, confirmed by sonography or laparoscopy.
We will also collect the following obstetric and perinatal complications:
Preterm birth: defined as birth of a fetus delivered after 22 and before 37 completed weeks of gestation.
Birth weight, including low birth weight (defined as weight <2500 g at birth), very low birth weight (defined as <1500 g at birth), high birth weight (defined as >4000 g at birth) and very high birth weight (defined as >4500 g at birth).
Large for gestational age (defined as birth weight >90th centile for gestation, based on standardised ethnicity-based charts) and small for gestational age (defined as less than 10th centile for gestational age at delivery based on standardised ethnicity-based charts).
Congenital anomaly (any congenital anomaly will be included).
Stillbirth: defined as fetal death occurring during late pregnancy (at 22 completed weeks of gestational age and later) or during childbirth.
Neonatal death: defined as neonatal death occurring up to 7 completed days after birth.
Data management and monitoring
The data collected for the trial will include routine clinical data such as demographic data, fertility history and ART records, which are verifiable from the medical record and questionnaire data. All participating researchers and physicians will be required to receive relevant training in practices and procedures related to the study beforehand and to pass the associated assessments. For purposes of confidentiality and anonymity when recording data on report forms, study participants will be identified by an appropriate code number consistent with the allocated intervention.
All data are collected at baseline and follow-up through a standard clinical electronic data collection system. Initially, all researchers and physicians will be required to keep accurate and verifiable source notes in the medical record relevant to each participant’s eligibility criteria. After the recruitment of eligible participants, trained assessors will be assigned responsibility for data entry. This will require log-on procedures to a secure data portal, protected by individual assessor ID. Data will be uploaded from medical records to electronic case report form with the personal trail ID of each participant. When the trial is close-out, all participant-identifiable data, such as consent forms, screening and identification logs will be stored in the investigator site files, accessible only to delegated members of the study team.
Sample size calculation
According to the literature18 and the data of our centre, live birth rates among women with DOR in the control arm were around 25.0%. The live birth rates are comparable after fresh and frozen cycles in our centre. Based on other studies within fertility care, and the opinions of gynaecologists and epidemiologists, we assume that the minimal clinically important difference to make TLSs preferable over conventional incubators would be 10.0%. To demonstrate this difference with the two-sided test, 5.0% alpha-error, 80% statistical power, and taking consideration of a possible withdrawn rate as 10%, the minimum numbers of participants we need to enrol for the study are 730. The ratio between the test and control group will be 1:1.
Statistical analysis
Baseline characteristics will be described by descriptive analysis, and the balance between the two arms will be assessed. For continuous variables, the normality test will be estimated using frequency histograms and the Shapiro test initially. Where the parameters are normally distributed, they will be presented as mean with SD. If the parameters are non-normally distributed, their medians and IQRs will be reported. For categorical variables, we will present the proportions of the two arms. Also, we will report the numbers of recruitment, participants lost to follow-up, protocol violation and other relevant descriptive data.
Data analysis of this trial will follow the intention-to-treat principle, which includes all randomised women in the primary comparison between the two groups who did not withdraw from the study. The per-protocol analysis may be conducted as a secondary analysis. The primary outcome, cumulative live birth rate, will be compared between the two groups using Pearson’s Χ2 test or Fisher’s exact test for unadjusted analysis. We will also compute the unadjusted risk ratio (RR) and its 95% CI. In the event of prominent imbalances of potential confounders between the two groups, we will perform multivariable Poisson regression or log-binomial model to compute adjusted RR and its 95% CI. Secondary outcomes will be compared between the two groups using a similar approach.
Where values of baseline characteristics are missing, we will perform analysis by excluding missing values, we will then perform multiple imputations to impute missing values and conduct subsequent analysis to estimate the robustness of the findings. For loss to follow-up and protocol violation, we will attempt sensitive analyses to explore the effect of these factors on the trial findings.
The primary outcome will be compared between the two groups within the following subgroups including different ages (<35 years vs ≥35 years), fertilisation (IVF vs ICSI), ET (fresh vs frozen) and COH protocols in which the effects on outcomes might be modified. Due to the concern over the multiplicity of subgroup analysis, we will place limited importance on subgroup findings.
All tests will be two-tailed, and differences with p value<0.05 will be considered statistically significant.
Safety
All observed or volunteered adverse events such as gestational diabetes, antepartum haemorrhage and hypertensive disorders of pregnancy, regardless of treatment group or suspected causal relationship to intervention, will be recorded and reported to an independent data and safety monitoring board (DSMB).
The investigator will inform subjects and the reviewing accredited medical research ethics committee if any adverse event occurs, where it appears that the disadvantages of participation may be significantly greater than was foreseen in the research proposal. The study will be suspended pending further review by the accredited medical research ethics committee unless suspension would jeopardise the subjects’ health. The investigator will take care that all subjects are kept informed.
Interim analysis
The DSMB will perform an interim analysis within 3 months after ET has been completed in the first 365 randomised participants. They will do so using the endpoint ongoing pregnancy, as data on live births will not be available. The interim analysis will be conducted using a two-sided significant test with the Haybittle-Peto spending function and a type I error rate of 5% with stopping criteria of p value<0.002 (Z alpha=3.0). The study could be stopped prematurely based on the advice of the DSMB.
Patient and public involvement
All aspects of this study (development of the research question, study design and conduct of the trial, interpretation of results and editing of the final manuscript for publication) are taking place independently of patients and public involvement. The results will be disseminated to participants by their physicians.
Ethics and dissemination
This trial has been approved by the Institutional Ethical Committee of Shanghai First Maternity and Infant Hospital (KS1958). All participants in the trial will provide written informed consent. The study will be conducted following the principles outlined in the Declaration of Helsinki and its amendments, following the Medical Research Involving Human Subjects Act, and using Good Clinical Practice. The results of this trial will be disseminated through conference presentations and peer-reviewed scientific journals.
Trial status
The proposed study dates are from 1 April 2019 to 31 March 2020. The recruitment in the study centre commenced in December 2019 and will continue until the required number of participants is achieved, anticipated until November 2021.
References
Footnotes
MC, YW, XH, WL, CS, ZM and AA contributed equally.
Contributors MC, WL and BWM—study concept and design. MC, YW, XH, CS, ZM, AA, LH, CT, KL, YF, ZC, PK, YG and WL—acquisition of data. MC, YW, XH, WL and BWM—analysis and interpretation of data. MC, WL and BWM—drafting of the manuscript. YW, XH, CS, ZM, AA, LH, CT, KL, YF, ZC, PK, YG, WL and XT—critical revision of the manuscript for important intellectual content. MC, YW, XH, WL and BWM—statistical analysis. BWM and XT—study supervision.
Funding This work was supported by the Science and Technology Commission of Shanghai Municipality (19411960500).
Disclaimer The funding bodies had no role in the study design, implementation, analysis, manuscript, preparation or decision to submit this article for publication.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.