Article Text

Abstract
Introduction Crohn’s disease is a chronic inflammatory disease of the gastrointestinal tract with an increasing incidence and prevalence worldwide. The early use of anti-tumour necrosis factor agents, such as infliximab, in patients with an aggressive form of Crohn’s disease has become part of routine practice. However, infliximab has limitations, and early surgery might benefit patients more. The objective of this study was to compare laparoscopic bowel resection with infliximab treatment in patients with moderately or severely active Crohn’s disease with respect to endoscopic remission. The laparoscopic bowel resection combined with infliximab treatment trial is the first randomised controlled trial to demonstrate if early surgery can improve the outcome of patients with Crohn’s disease with limited non-stricturing disease treated with infliximab.
Methods and analysis This is a randomised, open-label, controlled trial at Renji Hospital. In this study, a total of 106 adult patients aged 18–80 years with moderately or severely active and steroid-dependent or steroid-resistant Crohn’s disease of the distal ileum will be randomly assigned in a 1:1 ratio to the control and surgery groups. The primary outcome is 12-month endoscopic remission measured by the Simple Endoscopic Score for Crohn’s Disease in the control group and the Rutgeerts score in the surgery group. The secondary outcomes are clinical remission, surgery rate, quality of life, Crohn’s disease-related medical costs and Crohn’s disease-related morbidity. The patients will be followed up every 6 months after randomisation through intestinal magnetic resonance enterography and colonoscopy for either 3 years or until clinical remission.
Ethics and dissemination All participants will provide informed consent. The protocol has been approved by the Medical Ethical Committee of the Academic Medical Center in Shanghai (No KY2019-180). Results will be disseminated through peer-reviewed journals and scientific conference presentations.
Trial registration number ChiCTR2000029323.
- inflammatory bowel disease
- surgery
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study is a randomised controlled trial comparing laparoscopic bowel resection combined with infliximab treatment and infliximab alone for terminal ileitis in Crohn’s disease.
This study is the first to explore if early surgery can improve the outcome of patients with limited non-stricturing Crohn’s disease treated with infliximab.
Findings from this study will provide us with clinical evidence that early surgery is an effective therapy for patients with limited non-stricturing Crohn’s disease.
The study may take long time and much money.
The long-term effect of early surgery for patients with Crohn’s disease is unknown.
Introduction
Crohn’s disease (CD) is an idiopathic chronic relapsing inflammatory bowel disease (IBD) with an increasing incidence and prevalence worldwide.1 While CD affects the entire gastrointestinal tract, it most often affects the terminal ileum.2 In China, the current estimated incidence of CD is 0.51–1.09 cases per 100 000 persons.3 4 Although infliximab (IFX) has been proven to be effective in the induction and maintenance treatment of patients with moderately and severely active CD, intestinal resection is eventually inevitable in many patients.5
Medical treatment is still the primary choice for most patients with CD, considering the potential morbidity associated with surgery. With the availability of IFX, maintenance therapy after IFX-induced remission has gradually become the main clinical treatment for patients with CD.6 However, IFX has limitations, including loss of response, expensive medical consumption and high recurrence rates after discontinuation.5 7 8
The natural course of CD shows that disease progression is a one-way process from inflammation to stenosis and/or fistula.9 Surgical treatment is an important part of the treatment of CD. It has been reported that 20%–40% of patients with CD require surgery in the first year after diagnosis, and up to 80% require surgery at some time during their disease.10 When patients with CD have acute complications such as intestinal obstruction, fistula, perforation or stenosis, or when medical treatment is ineffective or intolerable, surgery is usually inevitable. And long-term studies have shown that after resection, 50% of patients will never require reoperation.11 Some evidence suggests that postoperative IFX treatment may lead to a lower recurrence rate (9.1%–24% within 1 year) compared with not using IFX postoperatively (81%–85% within 1 year).12–16 However, several studies have shown that preoperative use of IFX can lead to an increased risk of postoperative complications such as sepsis, abdominal abscess and anastomotic leakage.17 18
Some studies believe that early surgery, which refers to surgery performed shortly after the diagnosis of CD, may benefit patients with CD more.10 19 Surgical treatment can be used as an initial treatment for localised CD that is resistant to glucocorticosteroids or immunomodulators and requires starting with anti-tumour necrosis factor treatment at a higher level. Some evidence suggests that early surgery in patients with CD may lead to a longer time to clinical recurrence and a lower long-term reoperation rate (14% at 5 years) compared with later surgery (30% at 5 years).10 The ’Laparoscopic ileocaecal resection versus infliximab for terminal ileitis in Crohn’s disease’ (LIR!C) trial compared laparoscopic ileocaecal resection with IFX therapy for patients with Crohn’s terminal ileitis who failed conventional therapy, and through the study, researchers found that the early surgery and IFX groups had similar endoscopic remission at 1 year, while the surgery group had better general quality of life and less medical consumption.20 Therefore, compared with the initial IFX treatment, we hold the opinion that early surgery can further reduce the side effects of drugs, improve the quality of life and reduce medical expenses. Hence, this trial was designed.
Methods and analysis
Study design
The study was designed as a randomised, open-label, single-centre, controlled study at Renji Hospital, Shanghai Jiao Tong University. MY will conduct the operational processes and statistical analyses. The study will be conducted in accordance with the principles of the Declaration of Helsinki and ‘good clinical practice’ guidelines. The protocol has been approved by the Medical Ethical Committee of the Academic Medical Center in Shanghai (No KY2019-180) and registered in the Chinese Clinical Trial Registry. Prior to randomisation, informed consent will be obtained from all patients.
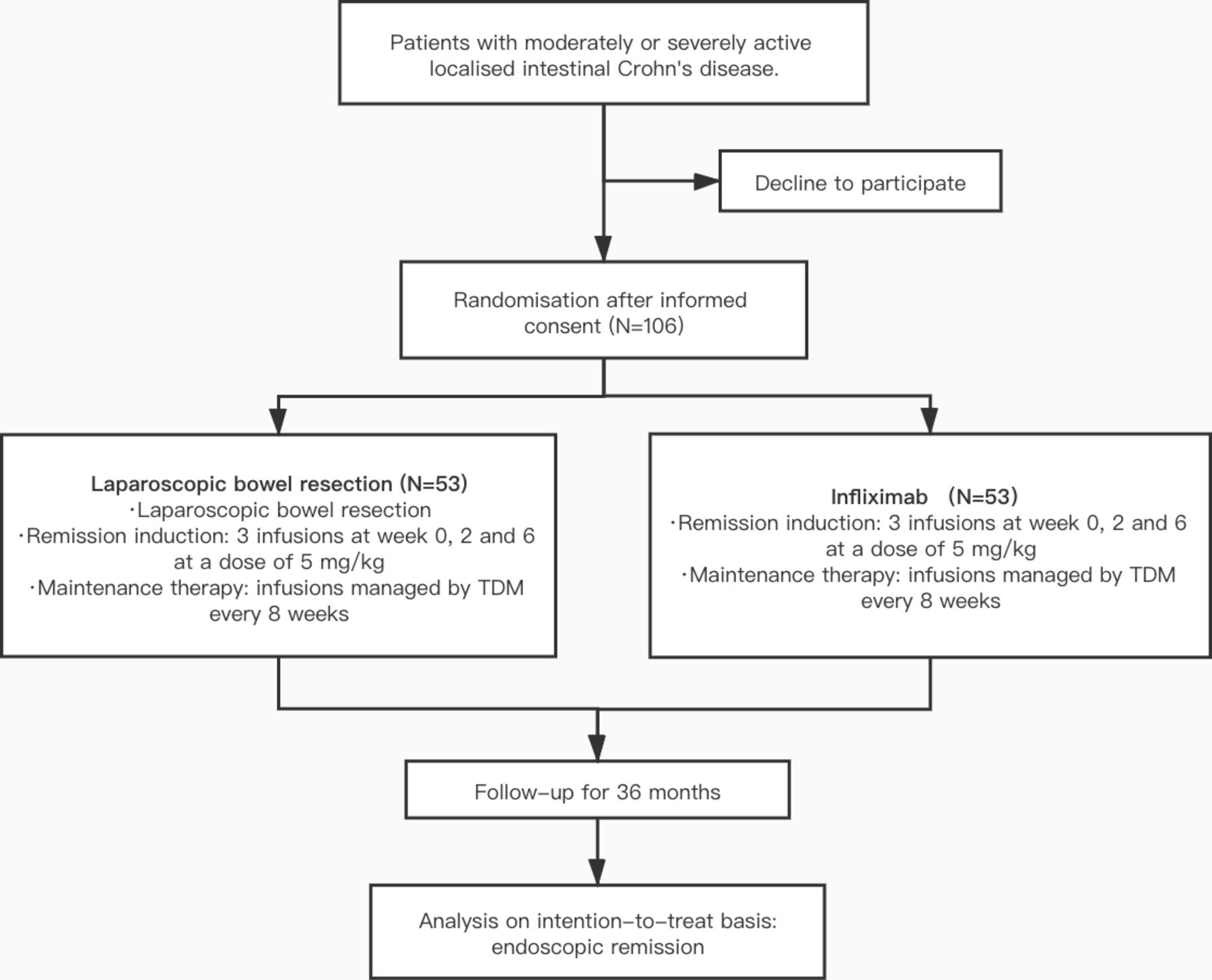
Eligible patients who give their consent to participate in the study will be randomly assigned in a 1:1 ratio to either treatment with laparoscopic bowel resection followed by IFX or IFX only. Both groups will be assessed for the outcomes at baseline, after the intervention and at the 3-year follow-up. Figure 1 shows the flow of participants in laparoscopic bowel resection combined with infliximab treatment (LaRIC) trial. During the follow-up period, general examinations and infectious screening will be scheduled before every IFX treatment. Patients are required to undergo intestinal magnetic resonance enterography (MRE) and colonoscopy every 6 months or until clinical remission. The end of the study is defined by the last follow-up of the last enrolled patient.

{kind=link}
Flow chart of participants in laparoscopic bowel resection combined with infliximab treatment (LaRIC) versus infliximab for terminal ileitis in Crohn’s disease trial. TDM, therapeutic drug monitoring.
This study protocol is reported in accordance with the Standard Protocol Items: Recommendations for Interventional Trials statement.21
Patient and public involvement
Patients and the public will not be involved in the development of the research question or in the design of the study. Patients will receive oral and written information about this trial; however, they will not be involved in the recruitment and conduct of the study. Besides, the burden of the intervention will be assessed by patients themselves. After signing an informed consent by the participants, they will be assessed for eligibility and data collection will begin.
Participants
Inclusion/exclusion criteria
Patients aged between 18 and 80 years with moderately or severely active CD (Crohn’s Disease Activity Index (CDAI) score >220 points) of the distal ileum, and steroid-dependent or steroid-resistant (failure of induction with steroids and maintenance with azathioprine or methotrexate) CD will be recruited in the outpatient IBD clinics of the participating medical centre. Patients who have previous intestinal surgery, penetrating or stricturing CD of the terminal ileum that probably requires dilatation or surgery, abdominal abscesses, affected small bowel segments longer than 30 cm and contraindications of IFX will be excluded. Patients with absence of intestinal inflammation on screening MRE will be excluded. All participants will be required to provide written informed consent.
Interventions
Infliximab
After group assignment, patients randomly allocated to the IFX treatment arm will undergo remission induction consisting of three subsequent infusions at weeks 0, 2 and 6 at a dose of 5 mg/kg and further maintenance infusions of IFX dosing will be managed by proactive therapeutic drug monitoring every 8 weeks.
In patients who experience disease recurrence during IFX treatment, intervals will be shortened to 6 weeks, and/or the dose level will increase to 10 mg/kg. Infusion reactions will be treated with 25 mg prednisolone and 2 mg clemastine intravenously prior to subsequent infusions.
IFX will be given without any comedication to patients with intolerance to the above-mentioned immunomodulating drugs or to patients with contraindications for the use of these drugs. Patients’ total blood counts and liver enzymes will be monitored 2 and 4 weeks after initiation of therapy and subsequently at 3-month intervals.
Laparoscopic bowel resection
Patients randomly allocated to surgery will receive a short course of steroids to reduce inflammation: prednisolone 40 mg intravenous dose (once a day) for 1 week, and 20 mg intravenous for 1 week. Once steroid therapy has been tapered to a dose of 20 mg/day, bowel resection can be performed.20
Surgery will be performed under general anaesthesia. Bowel resection will preferably be performed laparoscopically. A variety of techniques can be applied to perform laparoscopic ileocolic resection ranging from facilitated (laparoscopic mobilisation of the right colon followed by extracorporeal vascular ligation, bowel transection and reanastomosis) to total laparoscopic procedures (all steps are performed intracorporeally, including reanastomosis). Generally, three or four trocars will suffice. The minilaparotomy is preferably performed as an up and down transumbilical incision.
Patients will receive basal line assessments 3 weeks after the surgery. If their condition permits, patients will undergo IFX remission induction consisting of three subsequent infusions at weeks 0, 2 and 6 at a dose of 5 mg/kg. The subsequent IFX maintenance therapy is the same as in the IFX treatment arm.
Outcome measures
Primary outcome
The primary endpoint of the LaRIC trial is the 12-month endoscopic remission rate measured by colonoscopy using the Simple Endoscopic Score for Crohn’s Disease (SES-CD) or the Rutgeerts score in the surgery group. All endoscopies were reviewed by a single experienced reader. An SES-CD score of less than 6 and a modified Rutgeerts score of less than 2b are considered to indicate remission.
Secondary outcomes
The secondary outcomes of this study were as follows: (1) 12-month clinical remission, measured by the CDAI; (2) 12 and 36-month surgery rate; (3) quality of life measured by the Inflammatory Bowel Disease Questionnaire (IBDQ) and Short Form-36 Health Survey Questionnaire (SF-36); (4) CD-related medical costs (including surgery-related and medication-related expenses); and (5) CD-related morbidity. Patients were contacted monthly to assess infusion-related adverse events, postoperative complications according to the Clavien-Dindo classification (grade IIIa or worse with surgical, endoscopic or radiological intervention was considered clinically relevant), readmissions to the hospital and days of sick leave.22
Sample size calculation
The sample size calculation will be based on the 12-month endoscopic remission of patients with CD, our primary outcome. According to previous literature, the 12-month endoscopic remission of IFX is 51%–77%,23 24 and in surgery with postoperative IFX, the 12-month endoscopic remission is 76%–91%.12–16 It is expected that the patients in the surgery with postoperative group will achieve a greater endoscopic remission at the 12 months. It is anticipated by the research group that the 12-month endoscopic remission of 85% (surgery group) and 60% (control group), based on a two-tailed test of two independent means, with a significance (α) level of 0.05% and 80% power, a sample size of each group is required to have 48 participants. It was assumed that the loss rate of this study will be no more than 10%, and the minimum sample size for this study is 53 patients in each group for a total sample of 106 patients.
Randomisation
Eligible patients will be randomly allocated (1:1) to either the control group or surgery group with postoperative IFX treatment, using an interactive response system and integrated web response system to receive a treatment A or treatment B assignment. The allocation schedule was generated by the system vendor using a computerised random list generator. Patients, their treating doctors, the external data monitoring committee and sponsor representatives will not be masked to treatment assignment.
Blinding
No blinding will take place in this study. Eligible patients who have given informed consent will be randomised into either of the two groups.
Data collection and monitoring
Data collected will include patient demographics and disease phenotype according to the Montreal classification and therapy. Patient demographic data collected will include sex, body mass index, age at diagnosis, smoking habits, family history of IBD, concurrent medications and history of surgery.
Patients will be followed for a period of 36 months. Before every IFX treatment, patients will be required to undergo general examinations, including routine blood tests, liver and kidney function, C-reactive protein and erythrocyte sedimentation rate. The specific follow-up staff will also screen for infectious diseases such as hepatitis B virus, hepatitis C virus, treponema pallidum and mycobacterium tuberculosis. In addition to the general examinations and infectious screening before the use of IFX, disease activity will be assessed by calculating the CDAI. Patients will undergo faecal calprotectin testing, measurement of IFX and anti-antibody concentrations, chest CT, intestinal MRE and colonoscopy every 6 months or until clinical remission. Eight times during this follow-up period, patients will complete a set of questionnaires (the IBDQ and SF-36). Patients will complete the first set of questionnaires before randomisation, the next set at month 1 of therapy, the third set at month 6 and, after that, every 6 months. Patients will be contacted by telephone every month by a trial nurse to assess complications, additional interventions, readmissions, duration of hospital and intensive care stays, visits to the outpatient clinic, number of days of sick leave, social attendance and to ensure completion of the questionnaires. During the 1-year study period, if study endpoints are not observed, the patient will be considered lost to follow-up. If the loss rate is more than 10%, new patients will be recruited.
An electronic case record form (CRF) will include general patient data (sex, age, medical history), patient response to the questionnaires and data concerning the type of intervention, complications, mortality and the duration of hospital and intensive care stays.
The data in the study will be managed by the clinical trial staff who will be supervised by the principal investigator (PI). The PI will be responsible for ensuring the accuracy, completeness, legibility and timeliness of the data ported. Researchers will not be permitted to interpret records, and other researchers who are not involved in the patients’ treatment will assess the outcomes. Furthermore, to ensure intersite reliability, the PI will generate an operations manual (including the definitions for CRFs and guidelines for collecting data) before data collection begins.
The Clinical Research Institute of Shanghai Jiao Tong University Medicine School will be responsible for clinical data management. The Research Electronic Data Capture system will be used. Data in the electronic data capture system will be checked by a built-in algorithm that is monitored when modifications are required and traceable to source data. The trial sponsor will have access to the final trial data set.
The Data and Safety Monitoring Committee (DSMC), which is independent of the medical centre and investigators, will perform an ongoing review of predefined safety parameters and overall study conduct. The DSMC established by the Clinical Research Institutes, Shanghai Jiao Tong University School of Medicine, will comprise experts in clinical trials, biostatistics and intensive care medicine.
On all study-specific documents, other than the signed consent form, the participant will be referred to by a study code, not by name. All study-specific documents at the medical centre, including source document verification, informed consent, data quality, study drug reconciliation and the table of follow-up, will be monitored by the DSMC on several occasions during the study, independent of investigators and the sponsor. In addition, the study may also be audited by local or national regulatory authorities.
The study may be terminated or suspended at any time for the following reasons: (1) the request of the study management committee or a regulatory authority; (2) funding or management problems; or (3) drug-associated severe adverse events. The Clinical Research Institute of Shanghai General Hospital can make the final decision to terminate the trial and will supply reason(s) for the termination or suspension to the medical centre. Otherwise, the study is considered terminated on completion of all patient treatments and evaluations. When discontinuing the trial, all records shall be kept for confirmation.
Statistical methods
The statistical analysis will be based on intention-to-treat (ITT) data. The basic demographics of each group of subjects will be described, and the description will be based on the distribution of demographic data. If the measurement data are normally distributed, data will be summarised as the mean plus or minus the SD. Non-normally distributed data will be presented as medians and quartiles. The counting indexes will be expressed in the form of frequencies and ratios. The primary evaluation index response rate of the two groups will be analysed by the Χ2 test. Later, logistic regression will also be used, and the corrected response rate will be calculated by combining the relevant covariate calculations. The 12-month clinical recurrence rate and reoperation rate of other secondary indicators will be analysed by the Kaplan-Meier method and log-rank test. If the indicator is a count variable (adverse event), the Χ2 test or Fisher’s exact test will be used; if the indicator is a continuous measurement variable (hospitalisation cost), either the t-test or non-parametric test will be selected according to the distribution. For the overall quality of life-related scores and cost, a linear model will be used to adjust the overall quality of life-related scores over time and group based on the relevant baseline covariates. The statistical analysis will be performed using R analysis software (the latest version), and p≤0.05 will be considered statistically significant. We will perform retrospective per-protocol analysis in the future. IFX-resistant patients will be included into ITT analysis and they will be excluded in per-protocol analysis.
Discussion
In China, IFX treatment is indicated for patients with CD who are either steroid refractory or steroid dependent following treatment with steroids alone or in combination with immunomodulatory drugs such as azathioprine or methotrexate, or for patients with high-risk factors for CD that are hard to control. These guidelines are in concert with the European consensus on IFX treatment.25 However, the European Crohn's and Colitis Organisation (ECCO) also recommends the need for trials comparing IFX and surgery in its consensus statement, stating that ‘surgery is a reasonable alternative for patients with disease refractory to conventional medical treatment and should also be discussed’. The LaRIC trial aims to resolve this discussion and to provide an evidence-based best treatment strategy.
In the past, open surgery was the primary choice, but with the use of technology, advances in equipment and increased physician experience, laparoscopic surgery has become a safe and feasible surgical method in appropriate patients with CD. Several meta-analyses and Cochrane reviews showed no significant difference in any outcomes between laparoscopic and open surgery for small-bowel CD and reported fewer complications and incisional hernias in favour of the laparoscopic approach.26–28 The 2017 ECCO Surgical Guidelines note that laparoscopic bowel resection is the preferred surgical method for ileocaecal resection with a reasonable amount of surgical experience.29 Since CD is a benign disease, surgery does not require bare blood vessels or dissection of lymph nodes. Evidence suggests that laparoscopic surgery has significant advantages for ileocaecal resections in patients without a history of surgery. Compared with open surgery, although the operation time for laparoscopic surgery is prolonged, it improves clinical outcomes, including intestinal function recovery time, hospitalisation time and postoperative complications.30 Laparoscopic bowel resection may be more effective than IFX in the treatment of recurrent CD at the end of the ileum, improving quality of life and reducing costs.31
This trial compares medical therapy with (minimally invasive) surgery for CD of the intestine. The first analysis will provide short-term and medium-term results up to 1 year of follow-up. However, since long-term data from this cohort are especially important, we aim to develop a subsequent study to continue follow-up after the first year.
Considering the drawbacks of IFX treatment, bowel resection can be an equivalent alternative treatment, despite the small risk of serious surgical complications. These strategies have not been compared in a clinical trial to date. IFX treatment may be less effective than laparoscopic bowel resection when assessed by endoscopic remission. To improve the treatment of IFX, we use early surgery as induction remission. Surgery is more definitive, quick symptom relief and cure in some cases.32 Therefore, this study aims to answer the question if early surgery can improve the outcome of patients with CD with limited non-stricturing disease treated with IFX.
Ethics and dissemination
The protocol has been approved by the Medical Ethical Committee of the Academic Medical Center in Shanghai (No KY2019-180) and registered in the Chinese Clinical Trial Registry.
A manuscript will be prepared from these findings and submitted to peer-reviewed journal for publication. Findings will be presented at local and international conferences.
References
Footnotes
XH, TF and YY are joint first authors.
YQ, MZ and MY are joint first authors.
Twitter @Qiao_YQ
Contributors XH participated in the conception, design, writing of the study protocol and recruitment of participants. TF and YY participated in the design, writing and statistical analysis of the study protocol. YQ, MZ and SL were involved in the conception, design, the recruitment of participants and the acquisition of data. YS, RJ and YL were responsible for data collection. MY contributed to the conception, design, the revision and editing of the study protocol. All authors read and approved the final manuscript.
Funding The project is supported by grants from the National Natural Science Foundation of China (No 81702300 to MY, No 81873555 to MZ, No 81500408 to YY and No 81802308 to YL) and the Foundation and Frontier Projects of Chongqing Science and Technology Commission (No cstc2016jcyjA0079 to YY).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.