Article Text

Abstract
Objective In SUSTAIN 7, once-weekly semaglutide demonstrated superior glycated haemoglobin (HbA1c) and body weight (BW) reductions versus once-weekly dulaglutide in subjects with type 2 diabetes (T2D). This post hoc analysis investigated the impact of clinically relevant subject characteristics on treatment effects of semaglutide versus dulaglutide.
Design Analyses by baseline age (<65, ≥65 years), sex (male, female), diabetes duration (≤5, >5–10, >10 years), HbA1c (≤7.5, >7.5–8.5, >8.5% (≤58, >58–69, >69 mmol/mol)) and body mass index (BMI) (<30, 30–<35, ≥35 kg/m2).
Setting 194 sites; 16 countries.
Participants Subjects with T2D (n=1199) exposed to treatment.
Interventions Semaglutide 0.5 mg versus dulaglutide 0.75 mg (low-dose comparison); semaglutide 1.0 mg versus dulaglutide 1.5 mg (high-dose comparison), all subcutaneously once weekly.
Primary and secondary outcome measures Change in HbA1c (primary endpoint) and BW (confirmatory secondary endpoint) from baseline to week 40; proportion of subjects achieving HbA1c targets (<7%, ≤6.5% (<53, ≤48 mmol/mol)) and weight-loss responses (≥5%, ≥10%) at week 40; and safety.
Results HbA1c and BW reductions (estimated treatment difference ranges: –0.22 to –0.70%-point; –1.76 to –3.84 kg) and proportion of subjects achieving HbA1c targets and weight-loss responses were statistically significantly greater for the majority of comparisons of semaglutide versus dulaglutide within each subgroup category and, excepting glycaemic control within the low-dose comparison in HbA1c subgroups, this was irrespective of subgroup or dose comparison. Gastrointestinal adverse events, the most common with both treatments, were reported by more women than men and, with semaglutide, decreased with increasing BMI.
Conclusions Consistently greater improvements in HbA1c and BW with semaglutide versus dulaglutide, regardless of age, sex, diabetes duration, glycaemic control and BMI, support the efficacy of semaglutide across the continuum of care in a heterogeneous population with T2D.
Trial registration number NCT02648204.
- general diabetes
- diabetes & endocrinology
- clinical trials
Data availability statement
Data are available upon reasonable request. Individual participant data will be shared in datasets in a de-identified format, including datasets from Novo Nordisk-sponsored clinical research completed after 2001 for product indications approved in both the European Union and USA. The study protocol and redacted clinical study report will be available according to Novo Nordisk data sharing commitments. Data will be available permanently after research completion and approval of product and product use in the European Union and USA. Data will only be shared with bona fide researchers submitting a research proposal and requesting access to data, for use as approved by the independent review board and according to its charter. The access request proposal form and the access criteria can be found online. Data will be made available on a specialised Statistical Analysis System data platform.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The analysis was designed to provide insight on the influence of five of the most common and relevant patient-level factors from a clinical perspective.
The inclusion of comparator data allows for a more robust analysis and direct comparison of the differences in efficacy and safety of semaglutide versus dulaglutide across the subgroups and subgroup categories.
As the analysis is based on SUSTAIN 7 data alone, it may only be representative of the trial-specific patient population.
The relatively small number of subjects in each subgroup category is a limitation.
As this is a post hoc analysis of a randomised clinical trial, there are inherent limitations and, as such, the data should be interpreted with caution.
Introduction
The population of adults with type 2 diabetes (T2D) is heterogeneous, with varying clinical characteristics and comorbidities.1 The importance of considering this heterogeneity when making treatment decisions is emphasised in guidelines on the management of T2D,1 2 which recommend individualised patient-centred care considering the presence of comorbidities, including obesity, chronic kidney disease and cardiovascular disease.2 Some studies have attempted to identify clusters of patients according to their clinical characteristics and risk of complications, in the hope this might enable treatment to be more precisely targeted to those who are likely to benefit most.3 However, there is an ongoing debate about whether clustering or stratifying patients based on simple clinical characteristics is the most useful approach.4 5
Glucagon-like peptide-1 receptor agonists (GLP-1RAs) are an established treatment for T2D, recommended in current management guidelines.1 2 The efficacy and safety of two once-weekly (OW) subcutaneous medications from the GLP-1RA class, semaglutide and dulaglutide, were respectively investigated in the global phase 3a SUSTAIN (Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes)6–10 and AWARD (Assessment of Weekly AdministRation of LY2189265 in Diabetes)11–20 clinical trial programmes. Both drugs have also been investigated in large-scale cardiovascular outcomes trials.21 22 Post hoc analyses of the SUSTAIN and the AWARD trials have analysed patient subgroups across the continuum of T2D care.23–35 Such analyses showed consistent, clinically relevant reductions in glycated haemoglobin (HbA1c) and body weight (BW) with semaglutide across patient subgroups based on characteristics including age, baseline body mass index (BMI), baseline HbA1c, diabetes duration, race and ethnicity.23–26 28 Dulaglutide has also been shown to be efficacious across subgroups based on sex, age, duration of diabetes, beta-cell function, HbA1c, BW and BMI.29–35
In the phase 3b SUSTAIN 7 clinical trial, semaglutide and dulaglutide were compared head-to-head in subjects with T2D on background treatment with metformin.36 The trial showed superior reductions in HbA1c and BW with semaglutide versus dulaglutide, for both low-dose (semaglutide 0.5 mg vs dulaglutide 0.75 mg) and high-dose (semaglutide 1.0 mg vs dulaglutide 1.5 mg) comparisons.36 Although both semaglutide and dulaglutide have individually demonstrated efficacy across multiple patient subpopulations,23–27 29–35 it is as yet unknown whether the treatment differences observed in the SUSTAIN 7 trial are influenced by heterogeneity in the characteristics of the patients with T2D.
To evaluate whether clinically relevant patient characteristics (age, sex, diabetes duration, HbA1c and BMI at baseline) affected the efficacy and safety of semaglutide versus dulaglutide, post hoc analyses of data from the SUSTAIN 7 trial were performed.
Materials and methods
Trial design
The design of the SUSTAIN 7 trial has been previously reported.36 Briefly, this was an open-label trial in which subjects with uncontrolled T2D were randomised to receive semaglutide OW 0.5 mg or 1.0 mg, or dulaglutide OW 0.75 mg or 1.5 mg, as add-on to background treatment with metformin, and were followed throughout a 40-week treatment period. Semaglutide was administered subcutaneously via a prefilled injection device at one of two maintenance dose levels (0.5 mg or 1.0 mg OW), following a fixed-dose escalation regimen.36 Dulaglutide was administered subcutaneously in accordance with the regimen used in the phase 3 clinical trial programme (0.75 mg or 1.5 mg OW), without dose escalation.37
The trial was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice guidelines and the Declaration of Helsinki.
Patient and public involvement
The research question and endpoints, such as efficacy and safety, were informed indirectly by patients’ priorities, experiences and preferences, via input from clinicians during advisory board meetings. No patients were involved directly in the design, recruitment and conduct of the trial. Furthermore, the trial results were not directly disseminated to trial patients, but were publicly communicated and available via press release, trial portal and journal publication. In the trial, the burden of intervention was not assessed by the patients, nor were there any patient advisers involved.
Patient population
The inclusion and exclusion criteria for the SUSTAIN 7 trial are described in detail elsewhere.36 Key inclusion criteria were: diagnosis of T2D; age ≥18 years; HbA1c ≥7.0%–10.5% (53–91 mmol/mol). Key exclusion criteria were: estimated glomerular filtration rate <60 mL/min/1.73 m2; history of chronic or idiopathic acute pancreatitis; known proliferative retinopathy or maculopathy requiring acute treatment (determined by funduscopy/fundus photography performed within 90 days before randomisation according to local practice); screening calcitonin value ≥50 ng/L; personal/family history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2; acute coronary or cerebrovascular event within 90 days before randomisation; heart failure (New York Heart Association Class IV); and any of the following: myocardial infarction, stroke, or hospitalisation for unstable angina and/or transient ischaemic attack within 180 days of screening.36
Endpoints
The primary endpoint was change in HbA1c (%-point) from baseline to end of treatment at week 40 and the secondary confirmatory endpoint was change in BW (kg) over the same period. Predefined HbA1c treatment targets (proportion of subjects achieving HbA1c targets of <7% (53 mmol/mol) and ≤6.5% (48 mmol/mol)) and weight-loss responses (proportion of subjects achieving ≥5% and ≥10% weight loss) were also assessed.
The numbers of adverse events (AEs), serious AEs and AEs leading to premature treatment discontinuation were reported. Specific AEs of clinical interest, such as gastrointestinal (GI) disorders and hypoglycaemic events, were also evaluated.
Subgroup analyses
For this post hoc analysis, subjects were stratified into subgroups selected for potential clinical relevance: age at baseline (<65 years, ≥65 years), sex (male, female), diabetes duration at baseline (≤5 years, >5–10 years, >10 years), baseline HbA1c (≤7.5%, >7.5%–8.5%, >8.5% (≤58, >58–69, >69 mmol/mol)) and baseline BMI (<30 kg/m2, 30–<35 kg/m2, ≥35 kg/m2). The baseline BMI <25 kg/m2 subgroup category was also evaluated; however, due to the small number of subjects (representing less than 10% of the total trial population), these data are not included in the Results section, but are provided in the supplement.
Statistical analyses
The efficacy analyses were based on the full analysis set, comprising all subjects randomised and exposed to at least one dose of the trial product, using ‘on-treatment without rescue medication’ data (as randomised). Analysis of covariance was performed for each endpoint, including the interaction between treatment and subgroup as a factor. Multiple imputation was used to account for missing data. Specifically, using a sequential multiple-imputation approach, missing values for the underlying continuous assessments were imputed by treatment group, assuming missing data were missing at random, and based on a linear-regression model. A sequential conditional-regression approach was applied whereby missing observations at any post-baseline visits were imputed based on a linear-regression model and incorporating observations from previous visits including baseline. Binary endpoints were created and logistic-regression models run on the complete data set; inference was drawn using Rubin’s rule.38
Values for the mean change from baseline for HbA1c and BW were calculated, and the data are presented as mean and SE. Estimated treatment differences (ETDs) for the change from baseline in HbA1c and BW, and ORs for the proportions of subjects achieving HbA1c targets or weight-loss responses, both with 95% CIs, were also calculated for the low-dose (semaglutide 0.5 mg vs dulaglutide 0.75 mg) and high-dose (semaglutide 1.0 mg vs dulaglutide 1.5 mg) comparisons. To evaluate the evidence of heterogeneity of treatment effects across the clinical characteristics, a p value for interaction between treatment effect and subgroup categories was calculated for both dose comparisons in all subgroup analyses, without adjustment for multiplicity.
Safety analyses were based on the safety analysis set, which included all randomised subjects who were exposed to at least one dose of trial product, based on ‘as-treated’ data and summarised descriptively. Safety was assessed within each treatment arm (semaglutide 0.5 mg, dulaglutide 0.75 mg, semaglutide 1.0 mg, dulaglutide 1.5 mg) in each of the subgroup categories.
Analyses were conducted using SAS V.9.4. Baseline characteristics and AEs are provided as descriptive data only.
Results
Subject disposition and baseline characteristics
Baseline characteristics are summarised by treatment arm within each subgroup category (table 1; online supplemental tables 2–6). Subject characteristics were generally comparable across subgroup categories with some exceptions. In all treatment arms, diabetes duration was longer, and BW and BMI were lower in the elderly (≥65 years) subgroup compared with the non-elderly (<65 years) subgroup (table 1). Men were generally heavier but with a lower BMI, and had a longer diabetes duration than women (online supplemental table 2). In the diabetes duration subgroup categories (≤5 years, >5–10 years, >10 years), age increased with increasing diabetes duration and, in the semaglutide 1.0 mg treatment arm, BW and BMI decreased with increasing diabetes duration (online supplemental table 3). Across the baseline HbA1c subgroups (≤7.5%, >7.5–8.5, >8.5% (≤58, >58–69, >69 mmol/mol)), subjects in the semaglutide 0.5 mg treatment arm exhibited decreasing BW and BMI with increasing HbA1c (online supplemental table 4). In keeping with the distribution of subjects in the sex subgroup categories, there was a greater proportion of women versus men in the two highest BMI subgroups, and the proportion of Asian subjects was higher in the subgroup with the lowest BMI versus the subgroup with the highest BMI (online supplemental table 5). When compared with the other BMI subgroup categories, subjects with BMI <25 kg/m2 had the highest HbA1c levels, the highest proportions of men and Asian subjects and, as expected, the lowest BW (online supplemental table 6).
Supplemental material
Subject demographics and baseline characteristics by age
Glycaemic control and body weight changes
Missing observations in the efficacy analyses were mainly due to subjects who discontinued treatment or received rescue medication. At week 40, between 81% and 86% of subjects were on treatment without initiation of rescue medication in the four treatment arms (online supplemental figure 1).
Overall, the mean changes from baseline in HbA1c and BW (online supplemental figure 2) and the proportions of subjects achieving HbA1c targets of <7% (53 mmol/mol) and ≤6.5% (48 mmol/mol) and weight-loss responses of ≥5% and ≥10% (figures 1 and 2) were of greater magnitude with semaglutide versus dulaglutide treatment. This observation was confirmed by the ETDs for change from baseline (figure 3) and the ORs for proportions of subjects (online supplemental figures 3 and 4) which significantly favoured semaglutide in the majority of both the low-dose and high-dose comparisons within each subgroup category.

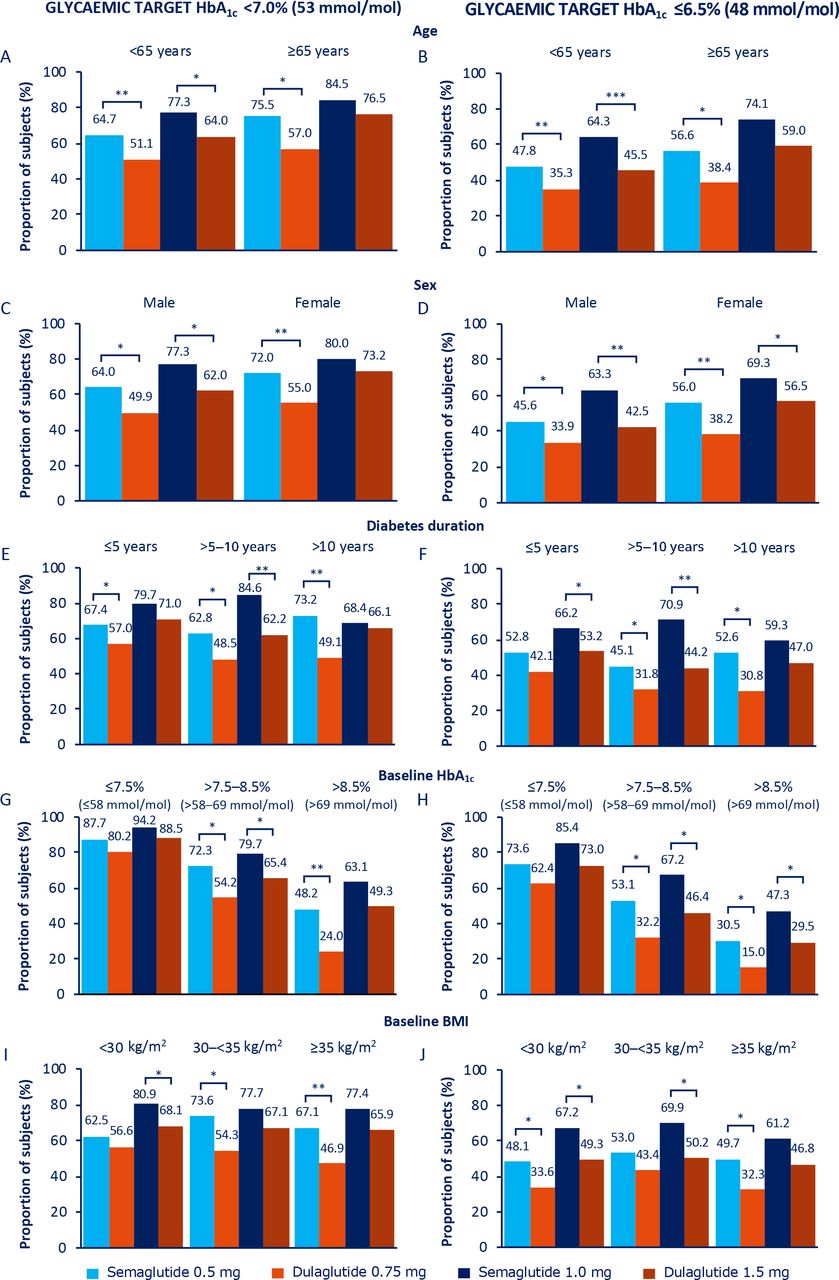
Proportion of subjects achieving HbA1c <7.0% (53 mmol/mol; A, C, E, G and I) and HbA1c ≤6.5% (48 mmol/mol; B, D, F, H and J) at 40 weeks. *P<0.05, **p<0.001, ***p<0.0001. Values are estimated proportions from ANCOVAs with multiple imputations using ‘on-treatment without rescue medication’ data from all randomised subjects exposed to at least one dose of trial product as randomised (full analysis set) obtained while on treatment and prior to onset of rescue medication. P values are based on ETDs; statistical analyses were not performed for change from baseline. ANCOVAs, analysis of covariances; BMI, body mass index; ETDs, estimated treatment differences; HbA1c, glycated haemoglobin.

Proportion of subjects achieving weight loss ≥5% (A, C, E, G and I) and weight loss ≥10% (B, D, F, H and J) at 40 weeks. *P<0.05, **p<0.001, ***p<0.0001. Values are estimated proportions from ANCOVAs with multiple imputations using ‘on-treatment without rescue medication’ data from all randomised subjects exposed to at least one dose of trial product as randomised (full analysis set) obtained while on treatment and prior to onset of rescue medication. P values are based on ETDs; statistical analyses were not performed for change from baseline. ANCOVAs, analysis of covariances; BMI, body mass index; ETDs, estimated treatment differences; HbA1c, glycated haemoglobin.

{kind=link}
{kind=link}
{kind=link}
Estimated treatment differences (ETDs) for change from baseline in HbA1c shown as %-points (A), HbA1c shown as mmol/mol (B) and body weight (C) at week 40 by age, sex, diabetes duration, HbA1c and BMI at baseline. *P<0.05, **p<0.001, ***p<0.0001; †P values represent the test for treatment by subgroup interaction. Values are ETDs (95% CIs) for semaglutide versus dulaglutide (low-dose comparison (semaglutide 0.5 mg vs dulaglutide 0.75 mg) and high-dose comparison (semaglutide 1.0 mg vs dulaglutide 1.5 mg)) from ANCOVAs with multiple imputations using data from all randomised subjects exposed to at least one dose of trial product who did not discontinue treatment or receive any non-investigational antihyperglycaemic treatment (full analysis set) while on treatment and prior to onset of rescue medication. ANCOVA controlled for baseline HbA1c (A and B) or body weight (C) and interaction between randomised treatment and subgroup. ANCOVA, analysis of covariance; BMI, body mass index; CI, confidence interval; ETD, estimated treatment difference; HbA1c, glycated haemoglobin.
For the individual analyses by subgroup, the findings were as follows:
Age at baseline (<65 years, ≥65 years): the proportion of elderly versus non-elderly subjects achieving glycaemic targets and weight-loss response of ≥5% was consistently numerically higher with both semaglutide and dulaglutide (figures 1A,B and 2A), despite elderly subjects having a lower baseline HbA1c and BMI than non-elderly subjects (table 1). Proportions of subjects achieving ≥10% weight loss were comparable between the two age subgroups for both treatment arms (figure 2B). Absolute changes in HbA1c and BW from baseline at week 40 by age are shown in online supplemental figure 2A,B.
Sex (male, female): reductions in HbA1c and BW were generally numerically greater in female than in male subjects (online supplemental figure 2C,D), as was baseline BMI (online supplemental table 2). This was reflected in the correspondingly greater proportions of female versus male subjects achieving the glycaemic targets and weight-loss responses (figures 1C,D and 2C,D).
Diabetes duration at baseline (≤5 years, >5–10 years, >10 years): comparatively smaller numeric reductions in HbA1c and BW were observed with semaglutide 1.0 mg in subjects with diabetes duration of >10 years versus ≤10 years, with no apparent differences observed in the other treatment arms (online supplemental figure 2E,F). A similar pattern was observed for the proportions of subjects achieving glycaemic targets and weight-loss responses in the semaglutide 1.0 mg treatment group (figures 1E,F and 2E,F).
Baseline HbA1c (≤7.5%, >7.5%–8.5%, >8.5% (≤58, >58–69, >69 mmol/mol)): with semaglutide 0.5 mg, and to a greater degree with semaglutide 1.0 mg, the magnitude of the mean reduction in HbA1c from baseline increased numerically with increasing baseline HbA1c; the converse was apparent for BW, whereby the amount of weight lost was less with increasing baseline HbA1c (online supplemental figure 2G,H). A similar though less apparent pattern was observed with dulaglutide (online supplemental figure 2G,H), and this was reflected in the proportions of subjects achieving glycaemic targets (figure 1G,H). Across baseline HbA1c subgroups, the greatest proportion of subjects achieving ≥5% weight loss was observed in those subjects receiving semaglutide 1.0 mg, particularly in the HbA1c subgroup categories of ≤7.5% (58 mmol/mol) and >7.5–8.5% (58–69 mmol/mol) (figure 2G). There were no other apparent differences across the subgroup categories regarding the proportions of subjects achieving weight-loss responses (figure 2G,H).
Baseline BMI (<30 kg/m2, 30–<35 kg/m2, ≥35 kg/m2): there were no apparent trends in glycaemic outcomes across the BMI categories for either dose comparison (figure 1I,J; online supplemental figure 2I). Mean reductions in BW for both semaglutide and dulaglutide increased numerically with increasing baseline BMI, with the greatest reductions in the ≥35 kg/m2 BMI subgroup category for all treatment arms (online supplemental figure 2J). There were no apparent trends in other BW outcomes across the BMI categories for either dose comparison figure 2I,J; online supplemental figure 2J), or when BW reduction was expressed as percentage change (online supplemental figure 5). Changes in the <25 kg/m2 BMI subgroup were largely consistent with those observed in the broader population (online supplemental figures 6 and 7).
Treatment–subgroup interaction effects
For each of the subgroups, analysis of the ETDs for the change from baseline in HbA1c in the age, sex, diabetes duration, baseline HbA1c and baseline BMI subgroups, the p values for the low-dose and high-dose comparisons were non-significant, except in the analysis of the HbA1c subgroups within the low-dose comparison (p<0.05 for the treatment–subgroup interaction effect) (figure 3A,B). The change from baseline in BW in the age, sex, diabetes duration, baseline HbA1c and baseline BMI subgroups was similar, with non-significant treatment–subgroup interactions for both dose comparisons (figure 3C). Similarly, treatment–subgroup interactions were non-significant for the analysis of the ORs for the proportions of subjects achieving glycaemic targets and weight-loss responses (online supplemental figures 3 and 4).
Safety outcomes
Overall, AEs were reported in more than half of subjects irrespective of the subgroup category (ranging from 55.3% (dulaglutide 0.75 mg; diabetes duration >5–10 years) to 80.6% (dulaglutide 1.5 mg; elderly)) and were generally more common with semaglutide 0.5 mg than with dulaglutide 0.75 mg, and less common with semaglutide 1.0 mg than with dulaglutide 1.5 mg. The proportions of subjects discontinuating treatment prematurely due to AEs were generally higher with semaglutide than with dulaglutide, and were primarily due to GI AEs (table 2; online supplemental tables 7–10).
Adverse events by age
GI AEs were the most frequently reported events, with generally higher rates with semaglutide 0.5 mg versus dulaglutide 0.75 mg, and dulaglutide 1.5 mg versus semaglutide 1.0 mg, across the subgroups and subgroup categories (ranging from 27.7% (dulaglutide 0.75 mg; diabetes duration >5–10 years) to 59.5% (dulaglutide 1.5 mg; HbA1c ≤7.5% (58 mmol/mol))), with nausea being the most common (ranging from 8.1% (dulaglutide 0.75 mg; male) to 29.5% (semaglutide 0.5 mg; female)) (table 2; online supplemental tables 7–10). Across the subgroup categories, more female than male subjects reported GI AEs overall, with GI AEs generally decreasing with increasing BMI in subjects treated with semaglutide (online supplemental tables 7 and 10). The highest proportion of GI AEs were reported by subjects with BMI <25 kg/m2 (online supplemental table 11).
Discussion
Given the heterogeneous profile of patients with T2D and the guidance for such differences to be considered when making treatment choices,2 3 this post hoc analysis of SUSTAIN 7 data assessed the impact of individual clinical characteristics on the effect of semaglutide versus dulaglutide treatment. The analyses indicate that the effect of semaglutide versus dulaglutide was not influenced by age, sex, diabetes duration, HbA1c or BMI at baseline, with the exception of the low-dose comparison for HbA1c in the baseline HbA1c subgroup, which showed increasing efficacy for semaglutide 0.5 mg versus dulaglutide 0.75 mg in subjects with increasing HbA1c at baseline.
This post hoc analysis supports the finding from the overall SUSTAIN 7 trial that semaglutide was superior to dulaglutide in reducing HbA1c and BW;36 the same was observed across each of the subgroups and within the various subgroup categories presented here.
This post hoc analysis also supports findings from similar subgroup analyses of SUSTAIN trials. An analysis of SUSTAIN 1–5 data showed greater reductions in HbA1c and BW with semaglutide versus comparators, and comparable efficacy in elderly subjects (a population often presenting with comorbidities) and non-elderly subjects, without an increased risk of hypoglycaemia.25 Similarly, analyses of pooled SUSTAIN data showed clinically relevant reductions in HbA1c and BW with semaglutide, regardless of baseline BW, HbA1c, diabetes duration, race and ethnicity.23–26 28
HbA1c reductions were greater with increasing baseline HbA1c for both semaglutide and dulaglutide in the present analyses, which has been observed with dulaglutide previously,29 31–33 as well as with liraglutide,39 lixisenatide40 and other antihyperglycaemic agents. Furthermore, a converse relationship between weight loss and baseline HbA1c levels was observed, whereby increasing baseline HbA1c was associated with greater reductions in HbA1c but a decreasing magnitude of weight loss. A similar pattern has been observed with liraglutide as an add-on to insulin treatment,41 with exenatide alone42 and with dulaglutide.31 32 These findings have relevance for clinical practice, indicating that there may be an effect with GLP-1RAs (and potentially other antihyperglycaemic therapies) in predicting treatment responses based on HbA1c levels.41 Conversely, a recent analysis of the AWARD trials found a weak positive correlation between HbA1c reduction and weight loss with dulaglutide.43 Several mechanisms, also associated with other antihyperglycaemic agents, may contribute to these results.44 Improved treatment-related glycaemic control is associated with decreased glycosuria,41 44 normalised protein turnover and a decreased catabolic effect,44 in addition to decreased energy expenditure and resting metabolic rate.44 As GLP-1RAs exhibit a glucose-dependent mechanism of action, the greater post-treatment reductions in HbA1c from a higher initial baseline HbA1c may contribute to the retention of glucose calories and, thereby, moderation of the achievable weight loss. In these analyses, greater weight loss was observed with increasing baseline BMI for both semaglutide and dulaglutide, aligning with what has been previously reported for semaglutide23 and dulaglutide.35 While percentage weight loss was also greater with semaglutide versus dulaglutide, the percentage change in weight loss was generally of a similar magnitude across BMI categories, indicating that the weight-loss pattern observed across the HbA1c subgroup categories may be associated with subjects’ baseline BMI. High BMI is associated with an insulin-resistant phenotype in some patients,3 and less weight loss is observed in patients with diabetes who are insulin resistant than in those with insulin sensitivity.45 However, clinically relevant reductions in BW were achieved for all BMI subgroup categories, and the magnitude of weight loss was comparatively greater for semaglutide than for dulaglutide. This is an important consideration for clinical practice, given the increasing interest in weight management as a key aspect of treatment for T2D.1
Analysis of the ETDs for change from baseline in HbA1c and BW and ORs for the proportions of subjects achieving HbA1c targets or weight-loss responses indicated a consistent effect of semaglutide versus dulaglutide across subgroup categories. These findings are aligned with previous analyses of subpopulations treated with GLP-1RAs, including semaglutide and dulaglutide, which also reported a non-significant impact of age, sex or diabetes duration on treatment effect,23–27 29–35 although weight loss tended to be greater in women than in men with dulaglutide,31 as was also observed in this analysis.
Consistent with the known class effect of GLP-1RAs,46 both semaglutide and dulaglutide reported relatively high levels of GI AEs. The rate of GI AEs was higher with semaglutide versus dulaglutide in the low-dose comparison; in the high-dose comparison it was higher with dulaglutide versus semaglutide.36 The proportions of subjects discontinuating treatment prematurely due to AEs were higher with semaglutide than with dulaglutide, which may be due to the higher levels of moderate GI AEs observed in the overall SUSTAIN 7 trial.36 The occurrence of some GIs AEs may be dose dependent, and nausea (and also vomiting for semaglutide) is usually transient with both semaglutide47 and dulaglutide;14 furthermore, the dose-escalation regimen approved for semaglutide has been shown to mitigate these AEs.47 In the subgroups in the present analyses, GI AEs were more frequent with dulaglutide 1.5 mg versus semaglutide 1.0 mg in elderly subjects with longer diabetes duration, and less frequent in subjects with HbA1c >8.5% (69 mmol/mol) and higher BMI. There were no other associations between subjects’ baseline characteristics with the incidence of GI AEs. Subjects who experience GI AEs, specifically nausea and vomiting, have greater weight loss compared with those who do not.23 48 While this hypothesised association might be considered an explanation for the observed greater weight loss with semaglutide versus dulaglutide in the low-dose comparison, a mediation analysis has previously shown that the direct effects of semaglutide on BW are the main contributors to weight loss with very little effect attributable to GI AEs.48 49 Our analyses support this finding as, overall, there were no clear trends between the incidence of GI AEs and the greater efficacy of semaglutide in terms of HbA1c reduction and weight loss versus dulaglutide.31 With semaglutide, there was a trend towards decreasing GI AEs with increasing baseline BMI, which has also been previously reported for the SUSTAIN 1–523 and the AWARD 1–650 trials, and may be due to differences in exposure–response levels associated with BW as has been demonstrated with semaglutide.51 Similarly, an analysis has shown that elderly patients with a lower BMI are more likely to experience side effects (including GI AEs) with dulaglutide than younger patients with a higher BMI.50 However, it is noted that this was a post hoc analysis in Japanese patients, with low event rates for some GI AEs, and so the results may not be generalisable to a wider diabetes population. In either case, a dose-escalation regimen may be beneficial.
A strength of the present analysis is the inclusion of comparator data, which allows for a more robust analysis and direct comparison of the differences in efficacy and safety of semaglutide versus dulaglutide across the subgroups and subgroup categories, and also the use of multiple imputation that helps to conserve randomisation. However, the post hoc nature of this analysis means there are inherent limitations and, as such, the data should be interpreted with caution. Also, as the analysis is based on SUSTAIN 7 alone, it may only be representative of the trial-specific patient population. A further limitation is the relatively small number of subjects in each subgroup category, which means that the findings should be interpreted with caution. Additionally, in the age subgroups, there was an imbalance in subject numbers (elderly vs non-elderly), with relatively few patients in the elderly subgroup (260; 22% of the analysis population). However, given the overall consistency of the age subgroup analyses, as well as the general limitations of these post hoc analyses, the difference in subject numbers between the age subgroup categories seemed to have had little or no impact. Furthermore, elderly subjects in previous pooled analyses of the SUSTAIN 1–526 and AWARD30 32 trials have demonstrated similar efficacy and safety, supporting the results obtained here.
Understanding the impact of heterogeneity in patient characteristics on treatment effects is important for clinical practice. This analysis provides insight on the influence of five of the most common and relevant patient-level factors from a clinical perspective and highlights semaglutide as an effective choice across these patient subgroups that are commonly encountered in clinical practice.
Conclusions
Semaglutide was associated with superior efficacy to dulaglutide across various clinically relevant patient subgroups that are commonly encountered in clinical practice, with a safety profile similar to other GLP-1RAs and in line with previously published data for semaglutide. The treatment effect for semaglutide versus dulaglutide did not appear to be influenced by age, sex, diabetes duration, HbA1c or BMI at baseline. Together with results from other studies and from experience in clinical practice, these findings support the efficacy of semaglutide across the continuum of care in a heterogeneous population with T2D.
Data availability statement
Data are available upon reasonable request. Individual participant data will be shared in datasets in a de-identified format, including datasets from Novo Nordisk-sponsored clinical research completed after 2001 for product indications approved in both the European Union and USA. The study protocol and redacted clinical study report will be available according to Novo Nordisk data sharing commitments. Data will be available permanently after research completion and approval of product and product use in the European Union and USA. Data will only be shared with bona fide researchers submitting a research proposal and requesting access to data, for use as approved by the independent review board and according to its charter. The access request proposal form and the access criteria can be found online. Data will be made available on a specialised Statistical Analysis System data platform.
Ethics statements
Ethics approval
The trial protocol (see online supplemental file) was approved by the central ethics committees (Eticka komisia, Nemocnica Svateho Michala, a.s., Bratislava, Slovakia and, for Portugal, Comissão de Ética para a Investigação Clínica) and by the institutional review boards and ethics committees at each participating centre (online supplemental table 1) and subjects provided written informed consent before trial-related activities commenced.
Acknowledgments
We thank all the trial subjects, investigators and trial-site staff members who were involved in the SUSTAIN trials; Charlotte Hindsberger and Thomas Hansen (both Novo Nordisk) for review and suggestions for revising the manuscript; and Stacy Carl-McGrath (AXON Communications) for medical writing and editorial assistance. This study and the associated trials were supported by Novo Nordisk A/S, Denmark. The funding sources contributed to the design and conduct of the trials, the analysis and interpretation of the data and the preparation, review and approval of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it was first published.
Contributors REP—conduct of trial, data collection, data analysis, data interpretation, manuscript preparation and approval of submitted version. VRA—conduct of trial, data collection, data interpretation, manuscript preparation and approval of submitted version. A-MC—data interpretation, manuscript preparation and approval of submitted version. IL—conduct of trial, data collection, data interpretation, manuscript preparation and approval of submitted version. JL—conduct of the trial, data collection, data interpretation, manuscript preparation and approval of submitted version. EY—data interpretation, manuscript preparation and approval of submitted version. AV—data interpretation, manuscript preparation and approval of submitted version.
Funding This work was supported by Novo Nordisk A/S. Editorial support was provided by Stacy Carl-McGrath PhD (AXON Communications), funded by Novo Nordisk A/S.
Competing interests The authors received no grants or funding for the writing of this article. In relation to the submitted work, RP reports grants from Novo Nordisk to his institution, AdventHealth, a nonprofit organisation, and VA also reports grants from Novo Nordisk to her institution.Outside of this work, RP reports speaker and consulting fees from AstraZeneca; consulting fees from Boehringer Ingelheim; consulting fees from Eisai, Inc.; consulting fees from GlaxoSmithKline; consulting fees from Glytec, LLC; consulting fees from Janssen; grants from Lexicon Pharmaceuticals; grants and consulting fees from Ligand Pharmaceuticals, Inc;, grants and consulting fees from Lilly; grants and consulting fees from Merck; consulting fees from Mundipharma; grants, speaker fees and consulting fees from Novo Nordisk; consulting fees from Pfizer; grants and consulting fees from Sanofi; grants, speaker fees and consulting fees from Takeda; and personal consulting fees from Sanofi US Services, Inc. Except for consulting fees in February 2018 and June 2018 from Sanofi US Services, Inc., RP’s services were paid for directly to AdventHealth. VA has received consulting fees (to her institution) from Adocia; grants (to her institution) and consulting fees (to her institution) from AstraZeneca/BMS; consulting fees from Becton-Dickinson; grants (to her institution) from Boehringer Ingelheim; grants (to her institution) from Calibra; consulting fees from Duke University; grants (to her institution) from Eisai; grants (to her institution) from Fractyl; grants (to her institution) from Janssen; grants (to her institution) and consulting fees from Novo Nordisk; grants (to her institution) and consulting fees from Sanofi; grants (to her institution) from Theracos; and consulting fees from Zafgen, all outside of the submitted work; and is the spouse of an employee of Merck Research Laboratories. AMC is an employee ofNovo Nordisk. In relation to the submitted work, IL reports grants to her institution from Novo Nordisk. Outside of the submitted work, IL has received consulting fees from AstraZeneca; consulting fees from Boehringer Ingelheim; grants from GI Dynamics; consulting fees from Intarcia; consulting fees from Janssen; consulting fees from Lilly; consulting fees from Mannkind; grants from Merck; grants from Mylan; grants from Novartis; grants and consulting fees from Novo Nordisk; grants from Pfizer; consulting fees from Sanofi; consultant fees from TARGETPharma; and consulting fees from Valeritas, all outside of the submitted work. JL reports being an investigator in clinical studies for Eli Lilly and Novo Nordisk outside of the submitted work. EY was, at the time of writing the manuscript, an employee and minor shareholder of Novo Nordisk. AV has received lecture/other fees and non-financial (clinical research) support from AstraZeneca; non-financial (clinical research) support from Amgen; lecture/other fees from Boehringer Ingelheim; lecture/other fees and non-financial (clinical research) support from Eli Lilly; lecture/other fees fromMerck Sharp & Dohme; lecture/other fees from Napp, lecture/other fees and non-financial (clinical research) support from Novo Nordisk; non-financial (clinical research) support from Novartis; and lecture/other fees and non-financial (clinical research) support from Sanofi, all outside of the submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.