Article Text

Abstract
Introduction Invasive aspergillosis is the most important cause of morbidity and mortality in patients with haematological diseases. At present, voriconazole is the first-line treatment for invasive fungal disease. The pharmacokinetic interindividual variability of voriconazole depends on genetic factors. CYP450 is involved in 70%–75% of total metabolism of voriconazole, mainly CYP3A4 and CYP2C19, with the remaining 25%–30% of metabolism conducted by monooxygenase flavins. CYP2C19 single nucleotide polymorphisms could explain 50%–55% of variability in voriconazole metabolism.
Materials and methods The main objective is to compare efficiency of pre-emptive voriconazole genotyping with routine practice. The primary outcome is serum voriconazole on the fifth day within the therapeutic range. The secondary outcome is the combined variables of therapeutic failure and adverse events within 90 days of first administration, associated with voriconazole. A total of 146 patients at risk of invasive aspergillosis who will potentially receive voriconazole will be recruited, and CYP2C19 will be genotyped. If the patient ultimately receives voriconazole, they will be randomised (1:1 experimental/control). In the experimental arm, patients will receive a dose according to a pharmacogenetic algorithm, including CYP2C19 genotype and clinical and demographic information. In the control arm, patients will receive a dose according to clinical practice guidelines. In addition, a Spanish National Healthcare System (NHS) point-of-view cost-effectiveness evaluation will be performed. Direct cost calculations for each arm will be performed.
Conclusion This trial will provide information about the viability and cost-effectiveness of the implementation of a pre-emptive voriconazole genotyping strategy in the Spanish NHS.
Ethics and dissemination A Spanish version of this protocol has been evaluated and approved by the La Paz University Hospital Ethics Committee and the Spanish Agency of Medicines and Medical Devices. Trial results will be submitted for publication in an open peer-reviewed medical speciality-specific publication.
Trial registration number Eudra-CT: 2019-000376-41 and NCT04238884; Pre-results.
- clinical pharmacology
- infectious diseases
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
The study will be randomised, with objective variables and not influenced by placebo effect (voriconazole serum level).
The effectiveness and safety related to serum voriconazole is well known.
For patients in this study, there is little risk compared with routine clinical practice.
The study is single-blinded.
Our results depend on the frequency of CYP2C19 polymorphism.
Introduction
Invasive aspergillosis is the most important cause of morbidity and mortality in patients with haematological diseases. The most recent epidemiological studies show an incidence of probable or proven invasive aspergillosis in patients with high-risk haematological diseases of 6%–11%.1 2 Mortality due to this complication in patients with acute leukaemia is 27%,1 and up to 40% if we include every malignant haematological disease.3 In recent years, great advances in the treatment of this pathology have been made, which have improved the prognosis of haematologic diseases. However, high morbidity and mortality associated with infectious complications continue to be a medical problem. As a result, improving the prognosis for invasive fungal disease has great scientific interest, with aspergillosis being the most prevalent form.
At present, voriconazole is the first-line treatment for invasive fungal disease (1-grade evidence, A-grade recommendation).4 5 It is a third-generation triazole antifungal with broad-spectrum activity. Achieving voriconazole serum levels in the therapeutic range during the first treatment week improves the prognosis of fungal infection and the tolerability of the treatment by reducing dose-dependent adverse effects (AEs).6 The AE reduction, associated with posology optimisation, decreases the amount of withdrawals of this antifungal drug.
Genetic variations could become markers that help us predict the pharmacological response depending on whether they are present and on their inter-relationship with other markers associated with clinical response. In recent years, several groups and organisations have published evidence assessment systems, guidelines, systematic reviews and evaluations for various drugs and pathologies; in some cases, genotyping prior to treatment with various drugs has been shown to be cost-efficient.7
Focusing on voriconazole, its pharmacokinetic interindividual variability depends on genetic factors.8 CYP450 is involved in 70%–75% of the total metabolism of voriconazole, mainly CYP3A4 and CYP2C19, with the remaining 25%–30% of metabolism conducted by monooxygenase flavins. CYP2C19 single nucleotide polymorphisms could explain 50%–55% of variability in voriconazole metabolism. Some 5%–17% of patients are ultrafast metabolisers (CYP2C19*17/*17), and approximately 25%–33% of patients are fast metabolisers (CYP2C19*1/*17). Both are associated with a high risk of not achieving therapeutic levels for invasive aspergillosis.9 10 Pharmacogenetics information oriented to dose adjustment is present in European dosing guidelines and in Food and Drug Administration recommendations, in which recommendations are given according to genotype.8 This information must be known by every physician in order to reduce adverse effects, improve effectiveness and thus raise patient compliance by reducing morbidity and mortality.
We propose a pragmatic clinical trial to evaluate the effectiveness and efficiency of a pre-emptive genotyping strategy of biomarkers related to the voriconazole response in patients with haematological disease who are at risk of suffering an infection susceptible to treatment with voriconazole.
Methods and analysis
Study design
VORIGENIPHARM is an acronym (VORIconazole PHARMacoGENetIcs) of the clinical trial, funded by the Spanish Health Research and Development Strategy. It is a phase IV pragmatic, multicentre, randomised, single-blind, parallel arm, centre-stratified clinical trial. A total of 146 patients at risk of invasive aspergillosis who will potentially receive voriconazole will be recruited, and CYP2C19 alleles will be genotyped. The patients who ultimately receive voriconazole will be randomised to receive the dose according to a pharmacogenetic algorithm, including the CYP2C19 genotype and clinical and demographic information, or according to clinical practice (figure 1).
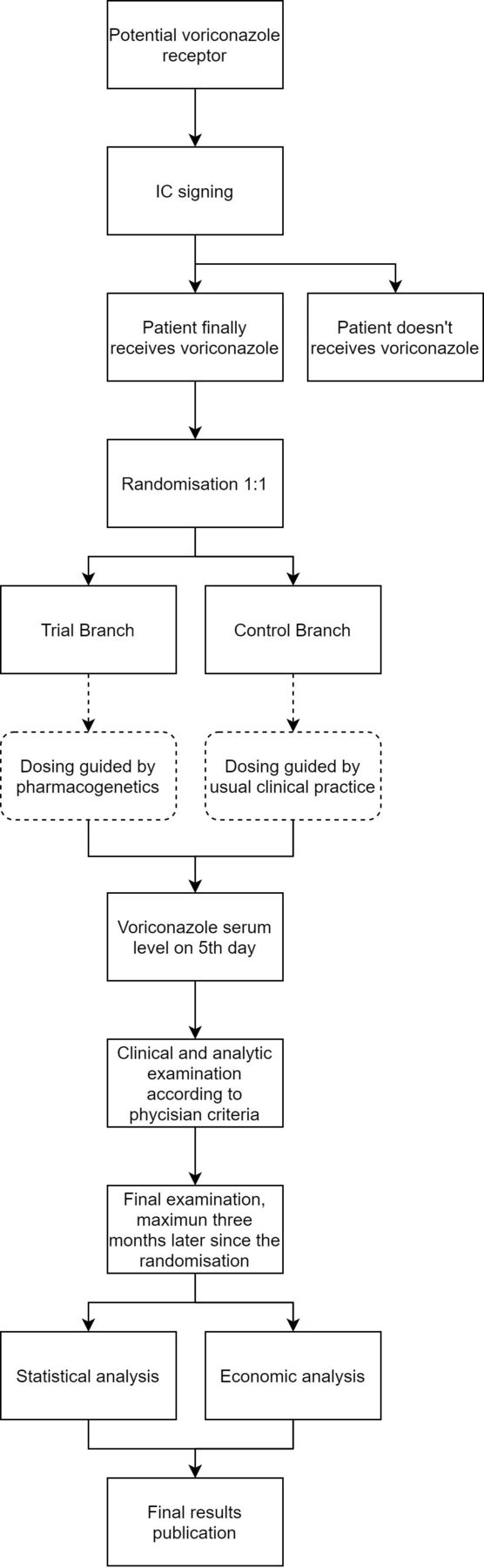
{kind=link}
Study flow chart. Proposed reporting of the flow of trial participants.
Study objectives
General objective
Evaluating the effectiveness and efficiency of a pre-emptive genotyping strategy for voriconazole in treatment and prophylaxis of Aspergillus fungal infections in patients with haematological diseases.
Primary objective
Evaluate the effectiveness of a pre-emptive genotyping strategy for voriconazole in achieving adequate therapeutic levels in haematological patients at risk of fungal infection compared with routine clinical practice.
Secondary objectives
Evaluate the safety of pre-emptive genotyping of voriconazole in haematological patients at risk of fungal infection, comparing it with routine clinical practice.
Evaluate the efficiency of this pre-emptive genotyping strategy. By mean of a cost-effectiveness analysis. Cost includes the mean total direct cost per patient, also including the cost of adverse events (AEs). The effectiveness will be measured as a combined variable of therapeutic failure described in the ‘Outcomes’ section.
Eligibility
The patient will be selected from La Paz University Hospital Paediatrics Hemato-Oncology Department, La Paz University Hospital Haematology Department, La Princesa University Hospital Adult Hematology Department and Gomez-Ulla University Hospital Adult Hematology Department. They will include children and adults who meet the following inclusion criteria:
Risk of developing invasive aspergillosis who are potentially eligible for treatment or prophylaxis with voriconazole:
Paediatric population: children undergoing haematopoietic stem cell transplantation, with acute myeloid leukaemia, as well as their relapses.
Adult population: patients diagnosed with acute leukaemia and those with long-term expected neutropaenia secondary to haematological disease and/or undergoing specific treatment (eg, aplastic anaemia and variants, myelodysplastic syndrome, solid organ or bone marrow transplantation), and patients who the clinician in charge considers might be at risk of developing fungal infection.
Accepting participation in the study by signing the informed consent (adult patients), or minor patients whose representative/legal guardian has willingly signed the informed consent. In the case of mature minors (12–17 years of age), in addition to the consent signed by the legal guardian, the minor’s assent shall be obtained.
The exclusion criteria are as follows:
Patients who for any reason should not be included in the study as assessed by the research team.
Patients who are not capable of understanding the information form and are unable to sign the informed consent document.
All patients eligible to receive voriconazole will sign an informed consent document for their participation in the clinical trial and for the collection of blood samples destined for genetic studies. They will also be asked for their consent to store an aliquot of their DNA for future studies (ie, mass sequencing). Patients identified in this phase will be randomised if and when they receive voriconazole.
Randomisation
Patients who meet all inclusion and no exclusion criteria, who have signed the informed consent and will receive voriconazole will be randomised, stratified by centres. The randomisation sequence was created using SAS V.9.4 statistical software (procedure ‘PROC PLAN’) with a 1:1 allocation. No randomisation seed was specified. The randomisation seed was generated taking the hour of the computer where the program was executed. Randomisation will be done centrally through the electronic CRF (MACRO) in order to conceal the sequence until interventions are assigned.
Masking
The study is single blind, and under no circumstance does the patient know the group to which they have been assigned. The medical researchers are unaware of the randomisation scheme. Although there is a risk of unmasking the patient, and the lack of masking for the physician could affect the evaluation variables, we believe that in this clinical trial is permissible since the primary variable is an objective one (voriconazole concentration), and a total masking is not feasible in a study using a pragmatic approach.
Outcomes
Primary outcome
Serum voriconazole levels in the range of 1–5.5 µg/mL on the fifth day, according to the British Society for Medical Mycology, which is a subrogated variable strongly related to effectiveness and safety, with high-level evidence.11
Secondary outcomes
A combined variable of therapeutic failure and AEs occurring within 90 days of first voriconazole administration.
Therapeutic failure is defined following European Organisation for Research and Treatment of Cancer (EORTC) consensus12:
Patients with probable or proven invasive aspergillosis: drug change or association because of poor clinical or radiological evolution of the disease.
Patients who received prophylactic treatment: the necessity of change because of probable or proven invasive fungal disease.
AE: dose-dependent drug AEs, including visual disturbances (eg, photopsia), skin reactions, neurotoxicity (eg, confusion and visual hallucinations) and QTc lengthening, requiring voriconazole withdrawal.
Costs by AE.
Cost savings by AE.
Quality-adjusted life years.
Study procedures
The study visits and procedures will be performed as shown in table 1.
Study visits planning and study procedures
Selection visit: after confirming the selection criteria, informing the patient and signing the informed consent document, a blood sample will be taken for genetic analysis. Patients who have not received voriconazole within 3 months of recruitment will not be considered for randomisation, but will be considered for economic evaluation. Patients who, in this 3-month period, receive voriconazole will continue to the randomisation visit.
Randomisation visit: the patient will be randomised to one of the two branches of the study:
Experimental group: based on the genetic study performed and the patient’s characteristics (age, weight, indication), the Pharmacogenetics Unit of La Paz University Hospital will indicate the dose to be administered. The dose will be based on the therapeutic individualisation protocol guided by pharmacogenetics, as agreed by all clinical services, and which includes the recommendations of the Clinical Pharmacogenetics Implementation Consortium guide (Moriyana, 2016) and the study by Hicks et al. In the experimental group arm, in cases of patients with rapid, ultrafast or slow metaboliser phenotypes, a determination of serum voriconazole concentrations at 48 hours (±24 hours) may be considered, which will make a dose adjustment possible if required.
Control group: no information will be provided and the procedure will be performed according to normal clinical practice, with clinical monitoring by the doctor in charge. In both cases, the request for serum voriconazole levels for subsequent dose adjustment will be recommended, as is standard clinical practice, in accordance with the recommendations of the British Society for Medical Mycology.6
Primary outcome evaluation visit: voriconazole concentrations will be measured on no later than the fifth day after the start of treatment (±1 day); if a dose adjustment is necessary, this will be performed according to the criteria of the attending clinician (control group) or the pharmacogenetic unit (experimental group); subsequent monitoring (determination of concentrations and dose adjustment) will be performed according to the criteria of the attending clinician in both arms.
Follow-up visits: while the patient is being treated with voriconazole, follow-up visits will be made according to clinical criteria, and the following information will be recorded:
Evaluation of potential voriconazole-related clinical AEs (see evaluation variables). This will be performed by asking the patient questions, performing a physical examination, routine tests (including liver enzymes) and an ECG.
Voriconazole levels, if requested according to medical criteria.
The type of consultation will be recorded: outpatient appointment, urgent episode, admission, readmission or phone call.
End-of-treatment visit: performed on the last day the patient receives treatment with voriconazole, including the following procedures:
A reason for the end of treatment will be indicated (withdrawal due to toxicity or therapeutic failure, or after completion of treatment according to clinical criteria of efficacy).
Evaluation of potentially voriconazole-related clinical AEs, as in follow-up visits.
If the patient continues voriconazole treatment beyond 3 months, the end-of-study visit will be made at that time.
End-of-study visit: the end-of-study visit will coincide with the end-of-treatment visit, except for those patients who are still undergoing treatment after 3 months. A safety analysis, ECG and assessment of AEs will be performed. Patients who continue treatment beyond this visit will be followed according to routine clinical practice.
Voriconazole genotyping and therapeutic drug monitoring
Genotyping will be performed at the Institute of Medical and Molecular Genetics at La Paz University Hospital using the self-designed SNP-array device (PharmArray V.2.), which makes genotyping of 180 relevant mutations possible for predicting the response to drugs. Among these mutations are the most relevant variants of CYP2C19, a gene that is related to serum levels of voriconazole, with a level of evidence 1A of CPIC: CYP2C19*2 (rs4244285), CYP2C19*3 (rs4986893) and CYP2C19*17 (rs12248560).
Serum voriconazole determinations will be tested at the Therapeutic Drug Monitoring Laboratory of the Clinical Pharmacology Service at La Paz University Hospital using an enzyme immunosensing technique on the Abbott ARCHITECT c4000 autoanalyser.
Data collection and outcome measures
A electronic case report form (eCRF) has been designed using MACRO Electronic Data Capture by Elsevier. This eCRF will include all the variables described and also concomitant medication and other clinical and demographic condition that could interfere with voriconazole. This system will anonymise patients, and the data will be transferred to a ‘*.csv’ file in order to analyse it with R software (V.3.5.2 or newer). To ensure the quality of the data, data management will be performed by the Spanish Clinical Research Network (SCReN). Data management plan have been approved by the principal investigator and the sponsor. Data collection forms will be included in the final report.
Sample size calculation
In routine clinical practice, 43% of patients have voriconazole serum levels in the therapeutic range on the fifth day of treatment. We expect this to improve to 68%, based on the article by Hicks et al.9 With a two-tailed type I error of 0.05% and 80% power, it is necessary to recruit 62 patients to receive treatment in both arms. Eighty-five out of every 100 patients recruited are estimated to receive voriconazole, we will need a total of 146 patients from whom we obtain informed consent. This calculation has been performed with the ‘power.prop.test’ package, from R V.3.2.0. The necessary time to recruit the whole group is estimated to be 2 years. We estimate 20% of the patients will be selected from the La Princesa University Hospital Adult Haematology Department, and 20% from the Gomez-Ulla University Hospital Adult Haematology Department. The rest of group will be recruited from La Paz University Hospital. Since it is a phase IV clinical trial including patients that are usually followed in the hospital, with pragmatic selection criteria, it is not necessary to plan any strategy for achieving adequate participant enrolment.
Statistical analysis
Frequency results will be expressed in absolute terms, as percentages and CIs. Continuous variables will be expressed as mean (SD) and median (range) according to Kolmogorov-Smirnov test of normality.
For the main dichotomous variable (level of voriconazole within the therapeutic range), a generalised logistic model will be used. After that, ‘elastic net’ techniques will be employed with the purpose of selecting variables so that they are not as rigid as in Lasso’s case. Variables with some degree of correlation between themselves will be accepted in the model, as in Ridge regression, aiming at the best possible predictive model. Finally, more advanced methods of statistical learning will be used to give the variables consistency by the method described above. Predictive ability will be calculated using receiver operating characteristic curves both at a specific time and over the course of the patient’s follow-up.
A survival analysis will be performed for the secondary variables to test the Cox proportional hazards model and the Weibull model, analysing the time until the first event, using Kaplan-Meier curves. A penalised Cox regression will be fitted to determine whether there are differences from the start of the target drug to the appearance of the event (secondary variable) between the control group and the experimental group, and finally, if possible the Weibull model will be used. A cross-validation approach will be used, dividing the sample into k subgroups. Later, a subgroup analysis will be performed taking into consideration the recruiting centre, calculating the a posteriori analysis power of every objective, given that this can be a source of hypothesis generation for future research lines.
The analysis of the primary variable of efficacy will be made on an intention-to-treat basis. It will include all the patients who are randomised, whether they have received the study treatment or not. The safety analyses will be held in the safety population, which includes patients who receive at least one dose of the drug during the study. No interim analysis or stopping rules will be carry out in this clinical trial.
A bilateral significance level of 0.05 and bilateral 95% CIs will be assumed. The statistical software R (R Core Team (2014)) will be used. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
Economic evaluation
A Spanish National Healthcare Service (NHS) point-of-view cost-effectiveness evaluation will be performed. Direct cost calculations for each arm will be performed.
Direct costs will be calculated for both alternatives by registering healthcare resources used during the follow-up and the unit cost per patient, as well as the costs incurred by the centres to implement the intervention.
The use of resources and their unit costs, such as doctor’s visits, medications and any interventions performed on those patients in the hospital during follow-up shall be recorded. Costs for each patient for each intervention group will be totalled. This calculation will allow us to compare the average cost per event avoided between the intervention group and the control group. The results will be expressed in cost per event and avoided events. As far as possible, the utility of each intervention should be calculated.
A univariate and a probabilistic sensitivity analysis will be performed to assess the uncertainty about the model and the unit costs, if they are obtained from a source other than the clinical centres.
Data monitoring
Coordination, management, monitoring, data management and statistical analysis of the study will be performed by the SCReN.
Auditing
During the progress of the study, audit visits can be conducted at the participating centres. The investigator will allow direct access to the source data/documents for monitoring, auditing, review by the ethical research committee and inspection by the health authorities.
Conflicts of interest
The funders and sponsor not interfere in the selection processes of the patients, analysis of the data and/or publication of the results, or any other process that might come into play with the results of the study. Funding will be independent of the results of the study. The Principal Investigator has ultimate authority over any of these activities.
Patient and public involvement
The development of the research question and outcome measures were based on the oncologists’ experience treating this profile of patients the desire to optimise voriconazole treatment. Patients and patient advisers were not involved in the design, recruitment or conduct of this study. The patients or their families will be notified of the study results in writing and verbally, and we will invite them to help us develop our dissemination strategy.
Access to data
This section is included in the data sharing plan.
Dissemination policy
Outputs from this study will include journal publications, conference presentations and community reporting. Outputs will not identify participants.
Ethical considerations
The researchers will adhere strictly to the provisions of this protocol and will complete the case report forms. The study will be performed according to the recommendations for clinical studies and the evaluation of drugs in humans, as contained in the Declaration of Helsinki (revised in successive world assemblies) and in the current Spanish and European legislation on clinical studies and patient data confidentiality. The study will follow the principles of Good Clinical Practice. This study has been approved by the Clinical Research Ethics Committee of La Paz University Hospital (Madrid, Spain) and by the Spanish Agency of Medication and Health Products.
This clinical trial has been classified by the Spanish Agency for Medicines and Healthcare Products as a ‘low-intervention clinical trial’. The additional diagnostic or monitoring procedures do not pose more than minimal additional risk or burden to the safety of the subjects compared with normal clinical.
No additional use of the system for compensation shall be required from the sponsor for low-intervention clinical trials. If any possible damage that could be suffered by a subject, resulting from the use of the investigational medicinal product in accordance with the protocol of that specific clinical trial, it is covered by the applicable compensation system already in place.
All protocol amendments will be evaluated by the Ethics Committee and the Spanish Agency of Medication and Health Products, following the principles of Good Clinical Practice and national legislation.
Discussion
Some important barriers have been detected to implementation of pharmacogenetics in usual clinical practice. The reasons detected in the literature include the difficulty of obtaining high-level evidence on genetic markers’ efficacy, effectivity and efficiency, and a lack of consensus. Another problem is the absence of pharmacogenetic techniques’ formation and its interpretation. We also have to consider the financial, logistic and legal limitations. Finally, there is no specific implementation strategy in clinical practice. There have been two proposed models for the implementation of pharmacogenetics biomarkers in usual clinical practice:
Case-to-case model: the decision is made individually and is based on the need for using a drug whose effectiveness or safety is modified by specific genetics variations. The limitations are the costs and the latency between sample extraction and the results obtained.
Pre-emptive genotyping model: this alternative assesses the patient’s genetic information at the very beginning, even before starting treatment. Currently, the development of arrays permits detection of a significant number of mutations in which therapeutic impact is probable or definite. Other authors have developed a similar strategy,11 12 for instance, St. Jude’s Hospital,13 as well as our group.14 Rasmussen-Torvik et al have proposed the eMERGE-PGx project, in which pre-emptive pharmacogenotyping is integrated with clinical history.15
In the case of voriconazole, several studies have linked CYP2C19 polymorphism to the response. In addition, some studies have shown that genotyping CYP2C19 is effective. The cost-efficiency of pretreatment genotyping in some drugs has been already demonstrated in some articles7; however, there is no evidence of voriconazole efficiency specifically in the Spanish NHS, or even whether voriconazole-related cytochrome genotyping is efficient in other countries.16
This pragmatic clinical trial is proposed by our group to evaluate effectiveness and efficiency of pre-emptive biomarker genotyping and its interpretation by a pharmacogenetic unit for patients with haematological diseases and a risk of fungal infection, who are susceptible to receiving voriconazole treatment. If our hypothesis is proven, this strategy will help us improve the prevention and treatment of this infectious complication. This trial will provide information about the viability of the translation of this strategy to routine clinical practice. In addition, our pre-emptive pharmacogenetic strategy, combined with therapeutic drug monitoring, has the potential to improve efficacy and safety, with a high level of evidence,6 lowering the likelihood of AE occurrences.
Acknowledgments
This Clinical Trial has been supported by Plataforma Española de Investigación Clínica y Ensayos Clínicos, SCReN (Spanish Clinical Research Network), funded by ISCIII-Subdirección General de Evaluación y Fomento de la Investigación, reserach project PT17/0017/0013. Plan Estatal de I+D+I 2013-2016. Plan Estatal de Investigación Científica y Técnica y de Innovación (2017-2020). Co-funded by European Regional Development Fund/European Social Fund "A way to make Europe"/"Investing in your future".
References
Footnotes
JMV and IGG contributed equally.
Author contributions AMB, AC and JFI conceived the study. JMV, IGG, FA-S, CA, ERG, JFI, AC and AMB designed the study. DB, RdlC, ME, ALG and MJO are responsible of the data collecting and management. JMV and IGG drafted this manuscript. AMB, AC and JFI critically revised the manuscript. JMV and IGG contributed equally. The final version of the manuscript was reviewed and approved by all authors.
Funding This clinical trial has been funded by the Instituto de Salud Carlos III (ISCIII), Minister of Innovation and Science of Spain in a competitive and public grant (Research Projects 2018, Spanish Health Research and Development Strategy). Project code: PI18/01322. (Co-funded by European Regional Development Fund/European Social Fund "A way to make Europe"/"Investing in your future")
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.