Article Text

Abstract
Background Although recent genome-wide association studies have identified several genetic variants contributing to the complex aetiology of multiple sclerosis (MS), expression and functional studies are required to further understand its molecular basis.
Objectives To identify genes and pathways with differential expression in MS.
Design The authors conducted a systematic review of seven microarray studies, in which expression in immune cells was compared between MS patients and controls. These studies include a previously unpublished study, which is described here in detail.
Results and conclusion Although in general the overlap between studies was poor, 229 genes were found to be differentially expressed in MS in at least two studies, of which 11 were in three studies and HSPA1A in four studies. After excluding the authors' unpublished experiment which may have been affected by certain confounding factors and inclusion of treated subjects, 135 genes were identified in at least two studies. The differentially expressed genes were significantly associated with several immunological pathways, including interleukin (IL)-4, IL-6, IL-17 and glucocorticoid receptor signalling pathways. 15 of the 229 loci have shown some association with MS in published genome-wide association studies (p<0.0001), including three loci with confirmed MS risk variants.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Article summary
Article focus
To identify genes showing differential expression in multiple sclerosis through genome-wide expression profiling in peripheral blood mononuclear cells.
To conduct a systematic review of genome-wide expression studies in multiple sclerosis in order to identify the most frequently reported genes.
To identify pathways associated with genes most frequently reported as differentially expressed in multiple sclerosis.
Key messages
The vast majority of all genes reported as differentially expressed were only identified in a single study.
However, 229 genes were reported as differentially expressed in MS to the same direction in at least two of the seven studies reviewed, 12 genes of which were in at least three studies.
After excluding our unpublished experiment. which may have been affected by confounding factors and inclusion of treated subjects, 135 genes were identified in at least two studies.
The differentially expressed genes were significantly associated with several immunological pathways, including the IL-4, IL-6, IL-17 and glucocorticoid receptor signalling pathways.
Strengths and limitations of this study
This is the first systematic review of genome-wide expression studies conducted in peripheral immune cells in multiple sclerosis.
Strict criteria were applied for inclusion of studies, and clearly underpowered studies with fewer than 10 cases or controls were excluded.
Many of the genes we found to be reported by at least two studies have interesting immunological functions and can be considered promising candidates for further studies.
However, the studies included should not be considered directly comparable owing to differences in samples, platforms and analyses methods used. In addition, the majority of these studies are small and should be viewed with some caution.
All studies were conducted in relatively heterogeneous cell populations, and some of the findings could therefore be explained by differences in numbers of different cell populations rather than differential transcriptional activity in MS.
Finally, our previously unpublished microarray study may have been affected by differences between the labs where the patient and controls samples were prepared for arrays, as well as by the higher mean age of controls. Our study also included four patients who had received immunomodulatory treatment at the time of sample collection.
Introduction
The aetiology of multiple sclerosis (MS) is complex and involves both genetic susceptibility and environmental factors. However, apart from the widely replicated association with human leukocyte antigen (HLA)-DRB1*1501, genetic risk factors have remained unknown until genome-wide association studies (GWAS), which have recently led to identification of common MS risk variants in over a dozen loci.1–6 Although further fine-mapping and functional studies are required in order to verify the causal variants and genes, a strong presence of immunological genes in these loci is evident. Expression and functional studies in immune cells can therefore elucidate the molecular mechanisms behind MS. Indeed, a number of studies have been conducted where genome-wide expression profiles in peripheral immune cells were compared between MS patients and unaffected controls. Together, these studies have reported a large number of genes with differential expression in MS. However, given that most of these studies have been conducted in small samples without replication, it is likely that many of the findings are false positives. Approaches are therefore needed to increase the probability of detecting the true signals from the vast number of reported genes. In order to extract the genes which are more likely to be true positives, we systematically reviewed results from seven microarray studies in MS, including our previously unpublished study. First, we identified genes which had been found to be differentially expressed in MS to the same direction in at least two studies. In order to further examine the potential role of these most frequently reported genes, they were analysed using pathway tools. Finally, we searched these genes for evidence of association by making comparisons with top results from recent MS GWAS.
Materials and methods
Samples in the Finnish microarray experiment
Twelve female patients fulfilling Poser's criteria for clinically definite MS were recruited through the Seinäjoki Central Hospital. Fifteen healthy unrelated female controls were obtained from the Finnish Twin Study on Ageing (FITSA). The mean age was 54.2 in patients and 71.6 in controls. One patient was receiving cortisone treatment, two patients received β interferon, and one patient was being treated with both β interferon and cortisone at the time of sample collection. All subjects had provided their informed consent. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood using BD Vacutainer CPT Cell Preparation Tubes (Becton, Dickinson and Company, Franklin Lakes, New Jersy), and cells were disrupted and RNA extracted with TRIzol Reagent (Invitrogen, Carlsbad, California). RNA was then purified using Rneasy Mini Kit (Qiagen, Hilden, Germany), and the sample quality was examined using BioAnalyzer (Agilent, Santa Clara, California). The study was approved by the Committee on Ethics of the Central Hospital of Central Finland and by the Helsinki University Hospital Ethical Committee of Ophthalmology, Otorhinolaryngology, Neurology and Neurosurgery (permit 192/E9/02) for FITSA and patient samples, respectively.
Sample processing and microarrays
Eleven patient samples were prepared for hybridisation on the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, California) according to the manufacturer's recommendations in our laboratory. In addition, one patient sample and technical replicates from two of the 11 patient samples were prepared according to the manufacturer's recommendations at the Helsinki Biomedicum Biochip Centre (BBC), where 15 control samples had been previously prepared. In brief, 1–2 μg of total RNA was converted to biotin-labelled cRNA using the Affymetrix HT One-Cycle cDNA Synthesis Kit and the HT IVT Labelling Kit. Fifteen micrograms of cRNA was then fragmented and hybridised for 16 h at 45°C, washed in Affymetrix Fluidics Station 450 and scanned with Affymetrix GeneChip Scanner 3000. Hybridisation, washing, staining and scanning were conducted using the same instruments for all samples. All arrays had a present call percentage >40 (42–47) and average background signal <50 (36–44).
Microarray data analysis
Raw intensity data files were imported to GeneSpring 7.3 (Agilent Technologies, Santa Clara, California) and GC Robust Multi-array Average (GC-RMA) normalised. We then applied filtering steps in order to exclude probe sets with low signal intensity and probe sets potentially affected by differences between our laboratory and the BBC. First, we excluded all probe sets with a GC-RMA normalised signal <50 in at least 20 of the 27 arrays (N=35 203). Second, we excluded probe sets where the technical replicates showed a >1.4-fold difference in both replicate pairs (N=1668). Finally, we excluded probe sets where the signal in all three MS arrays prepared at the BBC ranked among the four lowest or four highest among the MS arrays (N=2469), after which 15 273 probe sets remained for analyses. After filtering, we discarded the two replicate arrays prepared in our laboratory and used only the replicates prepared at the BBC for final analyses, which thereby included 12 MS arrays and 15 control arrays. In order to identify genes with differential expression in MS, we first applied the fold-change filter in GeneSpring 7.3 using 1.5 as threshold. For probe sets showing a ±≥1.5-fold difference in mean expression between MS patients and controls, we further determined non-parametric Mann–Whitney sum rank test p values with Benjamini–Hochberg correction for multiple testing. Probe sets with corrected a p value of ≤0.05 were considered to be differentially expressed. In order to annotate these probe sets, we compared the gene symbol obtained from GeneSpring 7.3 in October 2008 with the NetAffx annotation in December 2010 for each probe set (http://www.affymetrix.com/analysis/index.affx). If these were different, we first checked whether these were alternative identifiers for the same gene. If not, we obtained the probe set nucleotide sequences from NetAffx and performed a Blat search in UCSC Genome Browser (hg19) to identify the correct target gene (http://genome.ucsc.edu/cgi-bin/hgGateway). Probe sets which recognise only intronic sequences or map to several loci were excluded from the list of differentially expressed probe sets. The Gene Expression Omnibus (GEO) accession number for the microarray dataset is GSE21942 (http://www.ncbi.nlm.nih.gov/geo/).
Pathway analysis
Pathway analyses were conducted using the Core Analysis option in the Ingenuity Pathway Analysis software (Ingenuity Systems, Redwood City, California). This option identifies canonical pathways associated with a given list of genes by calculating the Fisher exact test p value for the probability that association between this set of genes and a canonical pathway is explained by chance alone. In order to account for the fact that our input lists of genes were enriched for immunological genes and would therefore show association with immune-related pathways if compared with all genes, we restricted the analyses to genes expressed in immune cells by applying the Tissues & Cell lines filter in the analysis settings.
Systematic review of microarray studies in MS
We searched PubMed with keywords ‘multiple sclerosis microarray’ in November 2010 and obtained 156 records. These were complemented with two additional recent studies and studies identified through a review article.7–9 Based on title, abstract and, if required, full text, we first identified all studies which had been conducted in peripheral immunological cells using a microarray platform and compared expression profiles between MS patients and unaffected controls. This led to the exclusion of 144 studies, which were not MS-related expression studies or investigated effects of MS treatments, were conducted in animal models for MS, or were performed in MS brain biopsies rather than immune cells. We then reviewed the remaining studies in further detail and excluded six studies with fewer than 10 MS patients and/or controls, a study which did not identify any differentially expressed genes, a study where only genes involved in T-cell mediated cytotoxicity were included in the analyses and a study where the list of identified genes was not available in the publication or upon contact with the corresponding author. The stages of the selection process are depicted as a flow chart in figure 1. Of the remaining eight studies which are listed in table 1, only six were independent, because the two studies by Satoh et al15 16 and the studies by Achiron et al10 and Mandel et al13 had been conducted in the same set of patients and were from hereon considered to be single studies. After including our own unpublished experiment, we therefore had seven independent studies for the analysis. For each study, we listed all genes which had been reported to be differentially expressed in MS patients in comparison with healthy controls, and recorded whether their expression in MS was increased or decreased. The studies by Achiron et al10 and Mandel et al13 listed only a selected subset of the identified differentially expressed genes.10 13 The corresponding author was contacted in order to obtain the full lists of differentially expressed genes, but we received no reply, and we therefore only included the reported genes. All gene symbols were mapped to the Human Gene Nomenclature gene symbols, and genes that were not unambiguously linked to a single gene symbol using the information available were excluded.

Flow chart showing stages in selecting studies for systematic review. MS, multiple sclerosis.
Description of previous microarray expression studies included in the systematic review
Results
The previously unpublished microarray screen identifies 692 probe sets with differential expression in MS
We first analysed the data from our previously unpublished Finnish microarray screen and identified 692 probe sets, which showed a ≥1.5-fold difference in mean expression between 12 MS patients and 15 controls together with a non-parametric Mann–Whitney sum rank test p value of ≤0.05 after Benjamini–Hochberg correction (supplementary table 1). Three hundred and one probe sets showed increased expression, and 391 decreased expression in MS. Pathway analysis revealed that the differentially expressed genes were strongly associated with PI3K signalling in B lymphocytes (p=1.3E–06), and B cell development (p=2.6E–06) pathways. In addition, altered T cell and B cell signalling in rheumatoid arthritis, role of PKR in interferon induction and antiviral response, and production of nitric oxide and reactive oxygen species in macrophages pathways showed evidence of association with the differentially expressed genes (p<1E–04).
Systematic review identifies 229 genes reported in at least two studies
In order to perform a systematic review of expression studies in MS, we identified eight previous studies, which filled our selection criteria (table 1). Studies which had been conducted in the same set of patients were combined, and we therefore considered six previously published independent lists of differentially expressed genes. Together with our previously unpublished experiment, these studies identified 2017 unique genes with increased expression and 1860 with decreased expression in MS, of which 303 genes were reported to have both increased and decreased expression. However, this list is not comprehensive, as two of the studies did not provide full lists of the identified genes. Two hundred and twenty-nine of the genes were found to have differential expression in MS to the same direction in at least two studies (supplementary table 2) and will be referred to as ‘in silico replicated’ differentially expressed genes (DEGs). One hundred and eleven of these had increased expression, and 119 decreased expression in MS, with one gene (STK4) identified as both. Eleven of the 229 genes were differentially expressed in three studies, and one gene, HSPA1A, in four studies (table 2). We acknowledge that our unpublished experiment may have been confounded by technical differences in processing of the patient and control samples, as well as by the age difference between cases and controls. In addition, the study included four patients who had received treatment, which may also have had an impact on the results. After excluding our study, 135 of the 229 DEGs were identified in at least two independent studies. These are indicated in supplementary table 2.
Genes identified as up- or down-regulated in multiple sclerosis in at least three studies
Several immunological pathways are significantly associated with the in silico replicated DEGs
In order to explore whether the in silico replicated DEGs are associated with specific pathways, we applied again the canonical pathway analysis option in the Ingenuity Pathway Analysis software. We conducted pathway analysis on the 135 DEGs identified in at least two independent studies excluding our unpublished study, and on the 229 DEGs identified when including our study. Unsurprisingly, many of the 20 most significantly associated pathways are related to immunological functions (tables 3, 4), with the most significantly associated pathway in both analyses being the glucocorticoid receptor signalling pathway. Other top-rated pathways include molecular mechanisms of cancer, NF-κB signalling and several interleukin (IL) signalling pathways (IL-4, IL-6, IL-17).
Top 20 pathways associated with the 229 in silico replicated differentially expressed genes
Top 20 pathways associated with the 135 in silico replicated differentially expressed genes after excluding the Finnish experiment
Fifteen in silico replicated DEGs are suggestively associated with MS
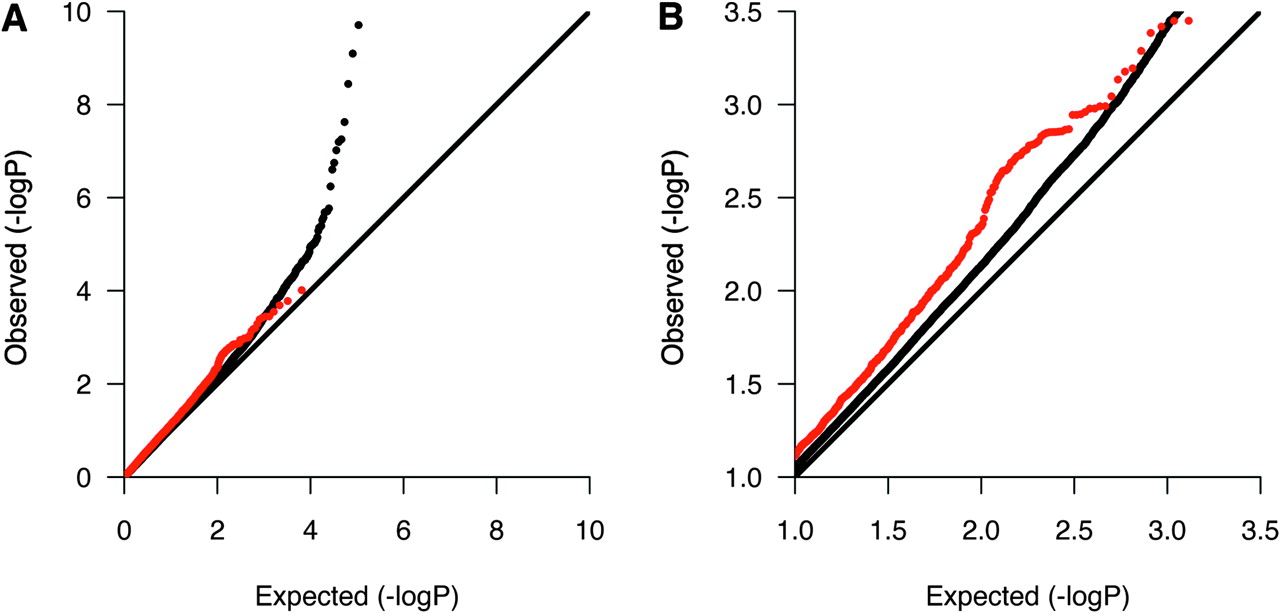
We then investigated whether the 229 in silico replicated DEGs showed any evidence for association in five previously published GWASs or in a recently published GWAS meta-analysis after excluding single nucleotide polymorphisms (SNP) in the human leukocyte antigen region (6p21–22).1–6 All association p values were available for the GWASs by Jakkula et al4 and International Multiple Sclerosis Genetics Consortium (IMSGC),1 and we therefore first examined the p value distribution of SNPs mapping within 100 kb of the 229 DEGs by quantile–quantile plots. Interestingly, these SNPs showed an overall enrichment of p values in the range 0.1–0.001 in the IMSGC GWAS (figure 2). However, we did not see any evidence of enrichment in the Finnish GWAS. We then reviewed the reported SNPs in the five published GWASs and the GWAS meta-analysis, and found 15 of the 229 in silico replicated DEGs (including regions 100 kb up and downstream) to contain suggestively associated SNPs (p≤0.0001) (table 5). The risk variants near three of the genes, CDK4, IL7R and TNFRSF1A, have been confirmed with genome-wide significance in GWASs or their follow-up studies (p≤5×10−8).1 3 5 17

Quantile–quantile plot for Cochran–Mantel–Haenszel p values in the International Multiple Sclerosis Genetics Consortium genome-wide association studies highlighting single nucleotide polymorphisms (SNP) in genes showing differential expression in multiple sclerosis. SNPs in 6p21–22 were excluded. Red dots represent SNPs mapping within 100 kb of in silico replicated differentially expressed genes. The remaining SNPs are represented by black dots. Plot A shows the entire p value distribution, and plot B is a close-up showing the enrichment of observed p values in the range 0.1–0.001.
Non-human leukocyte antigen single nucleotide polymorphisms (SNP) in silico replicated differentially expressed genes with p≤0.0001 in a genome-wide association study
Suggestively associated variant correlates with CXCR4 expression
To address the question, whether the above identified suggestively associated SNPs (p≤0.0001) mapping to 15 in silico replicated DEGs might contribute to the differential expression of these genes in MS, we first examined the genes using the mRNA by SNP Browser (http://www.sph.umich.edu/csg/liang/asthma/),18 19 which is a database of expression QTL (eQTL) SNP variants. However, none of the suggestively associated SNPs or their tagging SNPs (r2>0.8) correlated significantly with the expression of the corresponding DEG (p≤0.001). We also investigated the expression of these genes in risk allele carriers versus non-carriers in lymphoblastoid cell lines of 60 Centre d'Étude du Polymorphisme Humain (CEPH)-derived HapMap samples (CEU) (GEO dataset GSE5859)20 and obtained genotypes for the most strongly associated SNP, or SNPs if identified in several GWASs, from HapMap Release 24 (http://www.hapmap.org).21 In CXCR4, we tested rs882300, which was the most strongly associated CXCR4 SNP in a meta-analysis, instead of the two SNPs mapping within 100 kb of the gene. We did not identify any significant differences in expression between risk allele carriers and non-carriers after correcting for multiple testing. However, we found suggestive evidence for a higher expression of CXCR4 in carriers of the associated G allele at rs882300 (uncorrected one-sided p value=0.04, fold change=1.36) (figure 3), which is in concordance with the higher expression of CXCR4 observed in MS in our Finnish microarray study and three other studies, including two which were excluded from the systematic review owing to small sample sizes.11 22 23 This effect was also seen with the same probe set (217028_at) in our microarray data in a combined sample of 11 MS patients and 14 controls for which a genotype was available (fold change 1.19, one-sided Mann–Whitney test p value=0.06). However, no significant difference in expression levels was observed in two other probe sets measuring for the expression of CXCR4 in our microarray data. Both of these probe sets recognize exonic sequences, while 217028_at identifies 3′UTR in CXCR4.

{kind=link}
{kind=link}
{kind=link}
Box plot of CXCR4 expression and rs882300 genotype in 60 Centre d'Étude du Polymorphisme Humain (CEPH) lymphoblastoid cell samples.
Discussion
Despite extensive research and recent successes in identifying genetic risk variants predisposing to MS, the underlying molecular mechanisms still remain poorly understood. Given the suggested role of autoimmunity and predominance of immunological genes in loci associated with MS, genome-wide expression profiling in immune cells is a valid approach for further elucidating genes and pathways involved in the disease pathogenesis. Although several microarray experiments in MS have been conducted, and a large number of differentially expressed genes have been reported, in most cases the samples have been small, and replication has been lacking, making the findings difficult to interpret. While no obvious consistencies have emerged from these studies, there have not been any systematic attempts to evaluate the overlap between them. We therefore conducted a systematic review of seven microarray studies including our previously unpublished study and confirmed that the general overlap between the studies was indeed poor: only 229 of all 3574 (6%) genes reported to be differentially expressed in MS had been identified in at least two studies, and 94% were therefore unique to a single study. Only 12 of the 229 DEGs were identified in at least three studies. These include NEAT1, which encodes for a non-coding RNA and suppresses the expression of CIITA, an activator of genes within the major histocompatibility complex (MHC) class II locus.24 Interestingly, the most frequently reported gene, HSPA1A, which showed decreased expression in MS in four studies, is also functionally connected to the MHC: it encodes for a heat shock protein, which is likely to be involved in MHC class I and II mediated antigen presentation.25 The gene itself is located in the MHC class III region next to a highly homologous HSPA1B gene, and the measured expression levels may reflect the expression of both genes.
However, as is the case for expression studies in general, the studies are not directly comparable owing to differences in samples, sample sizes and platforms, as well as in criteria used for data quality control and for declaring differential expression. Furthermore, the majority of all reported genes came from the only two studies conducted in whole blood samples, and it is therefore not necessarily surprising that most of these genes were not identified in studies conducted in PBMCs or lymphocyte populations. Two studies also reported only a subset of the identified genes, and some studies were conducted using microarrays covering only a fraction of currently known human genes. However, perhaps the most likely explanation for poor overlap across studies is a high rate of false positives and low power to detect true differences in small samples. Recent GWASs conducted in large samples have proven that most of the early genetic associations reported in candidate gene studies of at most a few hundred individuals seem to have been false positives. Small samples may be even more problematic in expression studies, which are susceptible to noise introduced by technical and biological factors. Large studies are required, especially if the aim is to identify expression changes which are due to genetic disease risk variants because the effects of common genetic variants on gene expression are in most cases relatively modest, even in rather homogeneous cell populations,26 and in small samples the difference in risk allele frequency between cases and controls is not expected to be significant in the first place.
However, after excluding 13 genes in the MHC region, the 229 in silico replicated DEGs were enriched for variants showing a modest association in the IMSGC GWAS,1 suggesting that at least some of these genes are likely to play a causative role in MS rather than showing differential expression as a result of activation of immunological pathways secondary to MS. In addition, 15 of these genes have shown suggestive evidence for association with MS (p<0.0001) including CDK4, IL7R and TNFRSF1A, which are located in regions of genome-wide significance.1 3 5 17 These 15 also include CD40, which is associated with rheumatoid arthritis,27 and TNFAIP3, which is associated with coeliac disease, SLE, psoriasis and rheumatoid arthritis.28–31 TNFAIP3 was also one of the genes identified as differentially expressed in three studies, including our own experiment.11 15 16 It encodes for a zinc finger protein which inhibits nuclear factor (NF)-κβ activity and tumour necrosis factor (TNF)-mediated programmed cell death, and may therefore play an important role in regulating various immunological pathways.32 However, apart from CXCR4, the expression of the 15 DEGs did not correlate with the proposed risk variants in lymphoblastoid cell lines, although potential eQTL effects should be further investigated in other immune cells populations. In CXCR4, the associated SNP correlated modestly with expression when measured by a probe set representing the 3′UTR, which may indicate differential usage of alternative polyadenylation signals. Interestingly, CXCR4 promotes transendothelial migration of T cells in vitro together with its ligand, CXCL12,33 while CXCR4 and CXCR3 antagonists reduce the accumulation of CD4+ T cells in the CNS and inhibit EAE pathogenesis.34
We acknowledge that results from our previously unpublished experiment may have been affected by technical factors as well as by the age difference between cases and controls. The study also included four patients who had received treatment. We therefore reviewed the list of DEGs after excluding our study and found that 135 of the 229 DEGs were identified in at least two independent studies. Pathway analysis on both the 229 and 135 in silico replicated DEGs showed that they were highly associated with several immunological pathways. Interestingly, the identified interleukin signalling pathways (IL-4, IL-6 and IL-17) are primarily related to Th2 and Th17 cells rather than Th1 cells, which are thought to mediate MS.35–37 Further, IL-6 regulates the balance between regulatory T cell and Th17 cell differentiation together with TGF-β.38 Th17 cells have been linked with autoimmunity, and several studies have provided evidence for their role in MS and EAE,39 while regulatory T cells have been demonstrated to display loss of suppressive function in MS.40 Further studies are needed to investigate whether changes in expression of genes in these interleukin signalling pathways are causative or secondary to MS. We also saw a strong association with cancer-related pathways, which may suggest some common molecular mechanisms behind cancer and autoimmunity, such as dysregulation of apoptosis signalling. Finally, the pathway showing most significant association with the in silico replicated DEGs was the glucocorticoid receptor signalling pathway, which is a central regulator of inflammation. Although one could speculate that this may reflect the usage of corticosteroids as a treatment for MS, patients in the included studies had reportedly been untreated shortly prior to sample collection apart from our study where two patients had been treated with cortisone. This pathway was also the most significantly associated when our study, which included treated patients, was excluded. This would suggest that the regulation of endogenous glucocorticoid receptor signalling pathway may be disturbed in MS, which is in concordance with previous evidence of reduced glucocorticoid receptor binding affinity and sensitivity in lymphocytes in MS patients.41 42 Furthermore, mice producing an antisense RNA for the glucocorticoid receptor do not develop EAE.43 Interestingly, several of the genes in confirmed MS risk loci are connected to the glucocorticoid receptor signalling pathway, including STAT3, which was recently identified through a GWAS by our group4 and acts as a co-activator of glucocorticoid receptor signalling.44
In conclusion, we have performed the first systematic review of microarray studies in MS. In general, there was little overlap between the seven studies investigated, most likely owing to the small sizes of these studies. However, 229 genes were reported to be differentially expressed in MS in at least two studies. After excluding our unpublished experiment, which may have been affected by confounding factors and inclusion of treated subjects, 135 genes were identified in at least two studies. Of the 229 genes, 12 were reported in at least three studies, including TNFAIP3, which is associated with several other autoimmune diseases, and NEAT1 and HSPA1A, which are both functionally connected to MHC, the major MS susceptibility locus. Pathway analyses on the 229 and 135 DEGs provided support for the involvement of glucocorticoid receptor signalling and Th2, Th17 and regulatory T-cell-related interleukin signalling pathways in MS. Together with accumulating data from genetic association studies, our findings can be helpful in selecting genes and pathways for further functional studies in MS.
Acknowledgments
We are grateful to all MS patients and control subjects, who donated their samples to this study. We are deeply grateful to K Koivisto, for providing the patient samples, and to J Kaprio, for providing the control samples for the Finnish microarray study. L Puhakka, S Knaappila, M Suvela and A Nyberg are acknowledged for their technical assistance. The IMSGC consortium is acknowledged for GWA results. We wish to thank E Jakkula, V Leppä and I Surakka, for assistance in data handling and analyses. Finally, we owe our gratitude to L Peltonen-Palotie, for her supervision in this project.
References
Supplementary materials
Supplementary Data
Files in this Data Supplement:
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement - Manuscript file of format pdf
Footnotes
To cite: Kemppinen AK, Kaprio J, Palotie A, et al. Systematic review of genome-wide expression studies in multiple sclerosis. BMJ Open 2011;1:e000053. doi:10.1136/bmjopen-2011-000053
Funding This work was supported by the Sigrid Juselius Foundation, Helsinki University Central Hospital, the Academy of Finland Centre of Excellence in Complex Disease Genetics, the Neuropromise EU project (grant number LSHM-CT-2005-018637) and the Helsinki Biomedical Graduate School.
Competing interests None.
Ethics approval The study was approved by the Committee on Ethics of the Central Hospital of Central Finland and by the Helsinki University Hospital Ethical Committee of Ophthalmology, Otorhinolaryngology, Neurology and Neurosurgery (permit 192/E9/02) for FITSA and patient samples, respectively.
Contributors Sample handling and laboratory work: AK. Data analyses: AK. Manuscript writing: AK, JK, AP, JS. Approval of final manuscript: AK, JK, AP, JS.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The previously unpublished microarray expression dataset will be available through Gene Expression Omnibus (GSE21942).